
Structure and Catalytic Mechanism of Radical SAM Methylases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. RlmN and Cfr: How the Intriguing Chemistry of RSMases Was First Unveiled
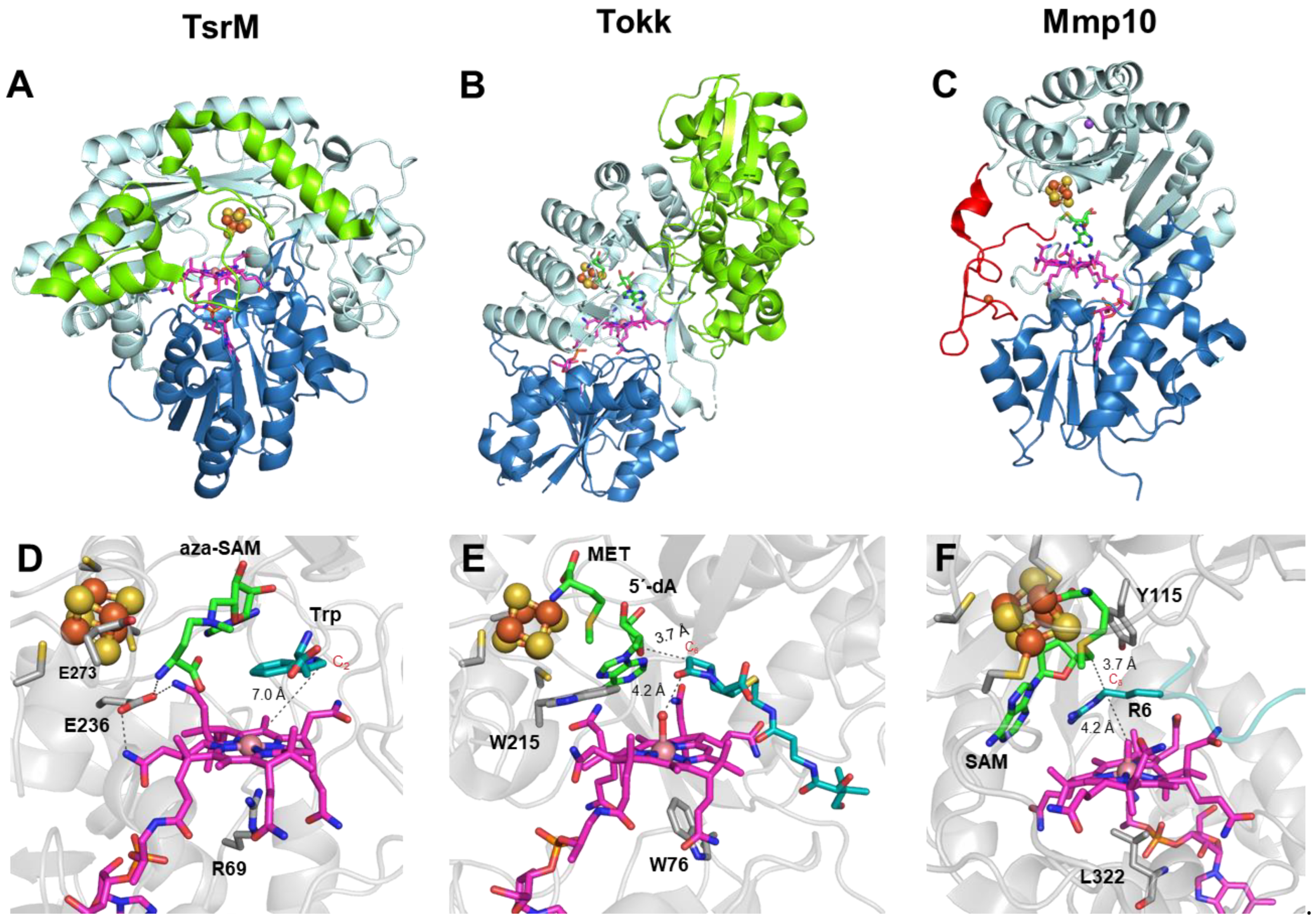
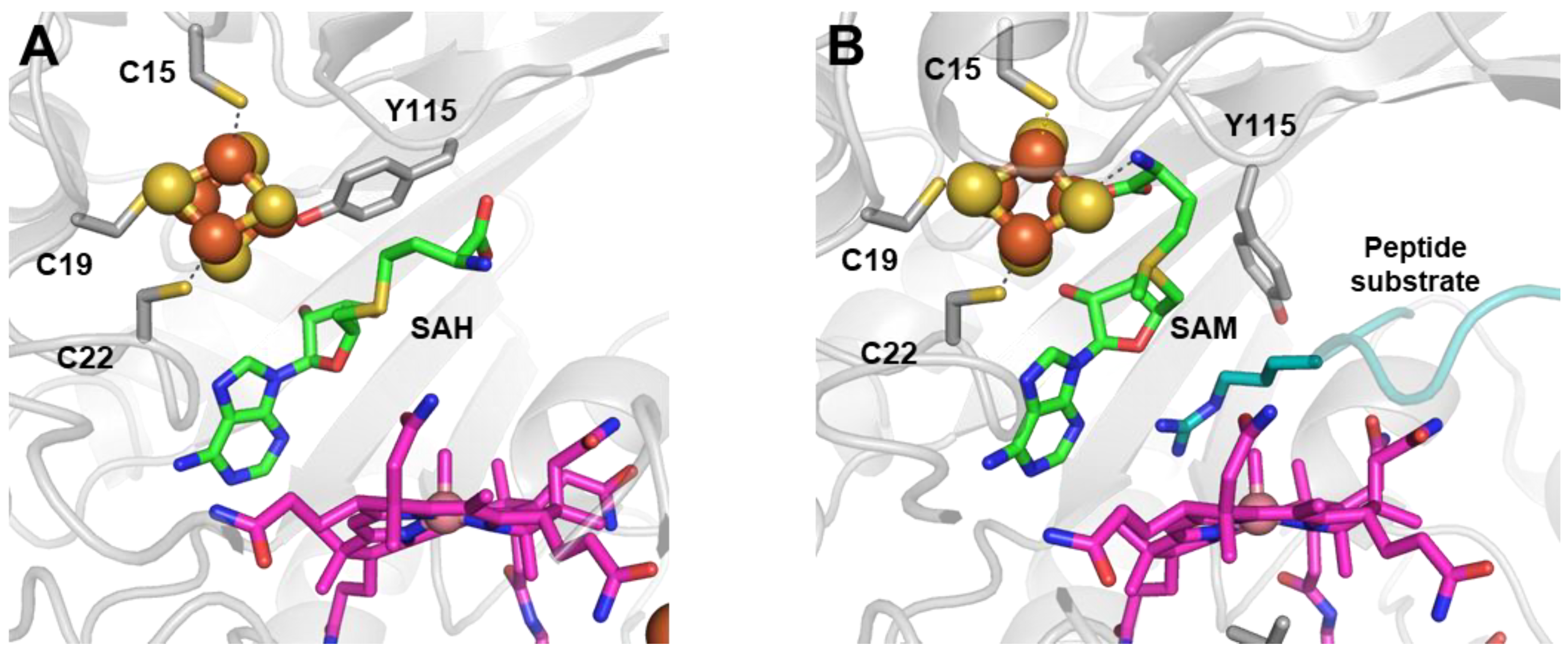
3. Cobalamin-Dependent RSMases: Similar Domains, Different Mechanisms
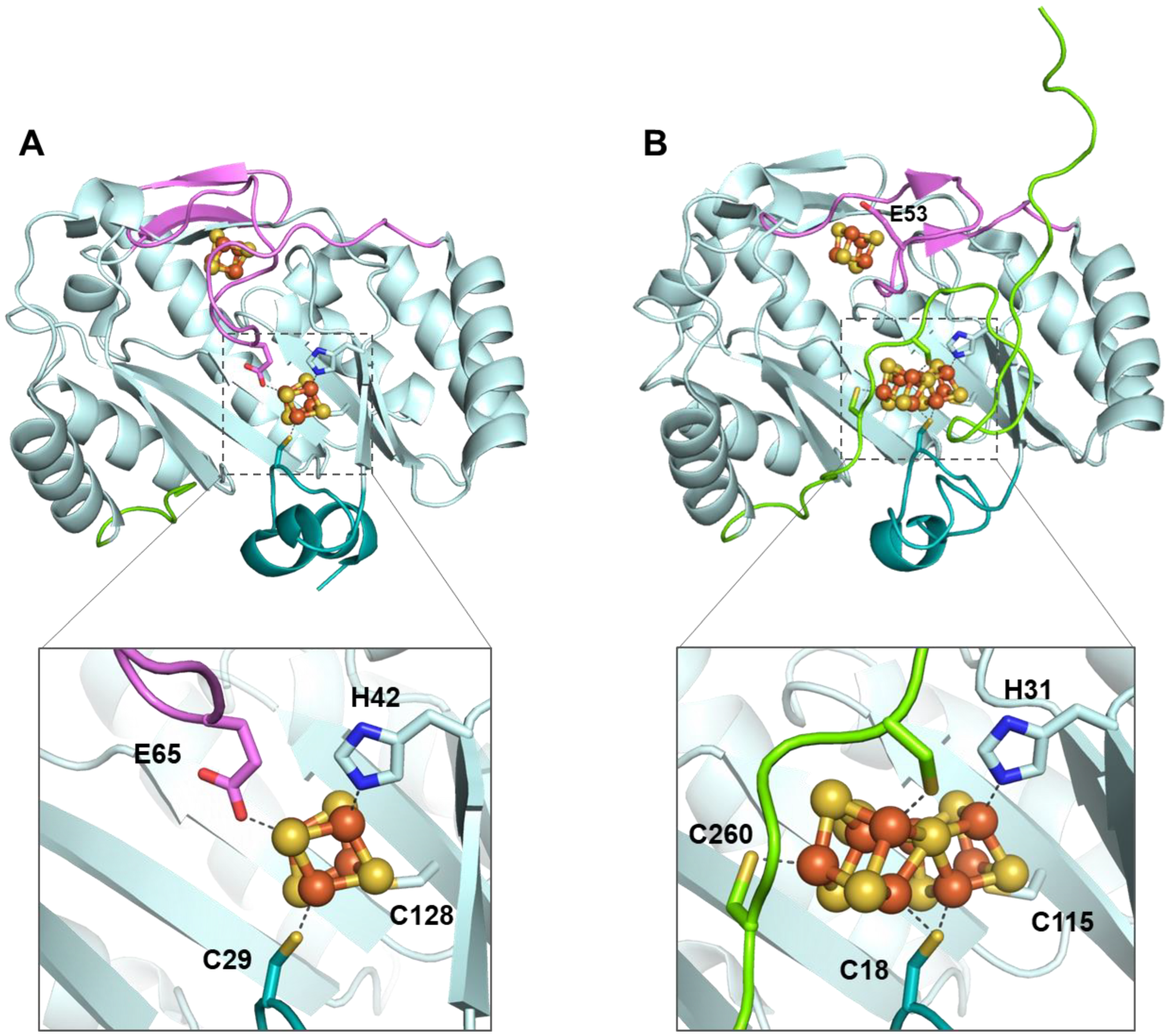
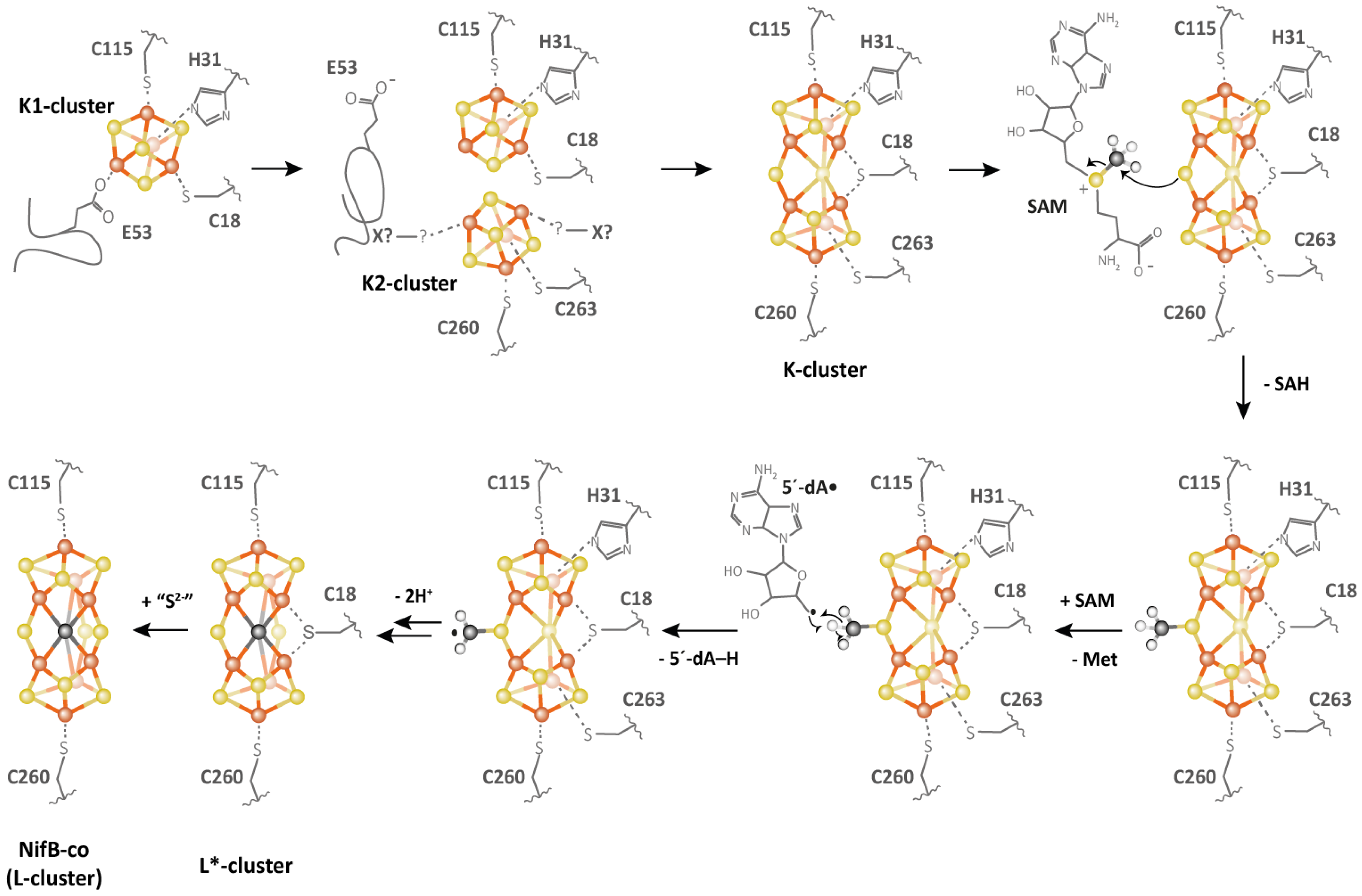
4. NifB: A Small RSMase That Goes beyond Methyl Transfer
5. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Struck, A.-W.; Thompson, M.L.; Wong, L.S.; Micklefield, J. S-Adenosyl-Methionine-Dependent Methyltransferases: Highly Versatile Enzymes in Biocatalysis, Biosynthesis and Other Biotechnological Applications. ChemBioChem Eur. J. Chem. Biol. 2012, 13, 2642–2655. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Arakawa, Y. 16S Ribosomal RNA Methylation: Emerging Resistance Mechanism against Aminoglycosides. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2007, 45, 88–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malanovic, N.; Streith, I.; Wolinski, H.; Rechberger, G.; Kohlwein, S.D.; Tehlivets, O. S-Adenosyl-L-Homocysteine Hydrolase, Key Enzyme of Methylation Metabolism, Regulates Phosphatidylcholine Synthesis and Triacylglycerol Homeostasis in Yeast: Implications for Homocysteine as a Risk Factor of Atherosclerosis. J. Biol. Chem. 2008, 283, 23989–23999. [Google Scholar] [CrossRef] [Green Version]
- Le, D.D.; Fujimori, D.G. Protein and Nucleic Acid Methylating Enzymes: Mechanisms and Regulation. Curr. Opin. Chem. Biol. 2012, 16, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Fontecave, M.; Atta, M.; Mulliez, E. S-Adenosylmethionine: Nothing Goes to Waste. Trends Biochem. Sci. 2004, 29, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Hegazi, M.F.; Borchardt, R.T.; Schowen, R.L. Alpha.-Deuterium and Carbon-13 Isotope Effects for Methyl Transfer Catalyzed by Catechol O-Methyltransferase. SN2-like Transition State. J. Am. Chem. Soc. 1979, 101, 4359–4365. [Google Scholar] [CrossRef]
- Iwig, D.F.; Uchida, A.; Stromberg, J.A.; Booker, S.J. The Activity of Escherichia Coli Cyclopropane Fatty Acid Synthase Depends on the Presence of Bicarbonate. J. Am. Chem. Soc. 2005, 127, 11612–11613. [Google Scholar] [CrossRef]
- Vidgren, J.; Svensson, L.A.; Liljas, A. Crystal Structure of Catechol O-Methyltransferase. Nature 1994, 368, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Trievel, R.C.; Beach, B.M.; Dirk, L.M.A.; Houtz, R.L.; Hurley, J.H. Structure and Catalytic Mechanism of a SET Domain Protein Methyltransferase. Cell 2002, 111, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Abaji, R.; Krajinovic, M. Thiopurine S-Methyltransferase Polymorphisms in Acute Lymphoblastic Leukemia, Inflammatory Bowel Disease and Autoimmune Disorders: Influence on Treatment Response. Pharm. Pers. Med. 2017, 10, 143–156. [Google Scholar] [CrossRef]
- Chen, L.; MacMillan, A.M.; Chang, W.; Ezaz-Nikpay, K.; Lane, W.S.; Verdine, G.L. Direct Identification of the Active-Site Nucleophile in a DNA (Cytosine-5)-Methyltransferase. Biochemistry 1991, 30, 11018–11025. [Google Scholar] [CrossRef] [PubMed]
- Kealey, J.T.; Gu, X.; Santi, D.V. Enzymatic Mechanism of TRNA (M5U54)Methyltransferase. Biochimie 1994, 76, 1133–1142. [Google Scholar] [CrossRef]
- Grove, T.L.; Radle, M.I.; Krebs, C.; Booker, S.J. Cfr and RlmN Contain a Single [4Fe-4S] Cluster, Which Directs Two Distinct Reactivities for S-Adenosylmethionine: Methyl Transfer by SN2 Displacement and Radical Generation. J. Am. Chem. Soc. 2011, 133, 19586–19589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H. S-Adenosylmethionine-Dependent Alkylation Reactions: When Are Radical Reactions Used? Bioorg. Chem. 2011, 39, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Sofia, H.J.; Chen, G.; Hetzler, B.G.; Reyes-Spindola, J.F.; Miller, N.E. Radical SAM, a Novel Protein Superfamily Linking Unresolved Steps in Familiar Biosynthetic Pathways with Radical Mechanisms: Functional Characterization Using New Analysis and Information Visualization Methods. Nucleic Acids Res. 2001, 29, 1097–1106. [Google Scholar] [CrossRef] [Green Version]
- Oberg, N.; Precord, T.W.; Mitchell, D.A.; Gerlt, J.A. RadicalSAM.Org: A Resource to Interpret Sequence-Function Space and Discover New Radical SAM Enzyme Chemistry. ACS Bio Med Chem Au 2022, 2, 22–35. [Google Scholar] [CrossRef]
- Nicolet, Y. Structure–Function Relationships of Radical SAM Enzymes. Nat. Catal. 2020, 3, 337–350. [Google Scholar] [CrossRef]
- Broderick, J.B.; Duffus, B.R.; Duschene, K.S.; Shepard, E.M. Radical S-Adenosylmethionine Enzymes. Chem. Rev. 2014, 114, 4229–4317. [Google Scholar] [CrossRef]
- Vey, J.L.; Drennan, C.L. Structural Insights into Radical Generation by the Radical SAM Superfamily. Chem. Rev. 2011, 111, 2487–2506. [Google Scholar] [CrossRef] [Green Version]
- Holliday, G.L.; Akiva, E.; Meng, E.C.; Brown, S.D.; Calhoun, S.; Pieper, U.; Sali, A.; Booker, S.J.; Babbitt, P.C. Atlas of the Radical SAM Superfamily: Divergent Evolution of Function Using a “Plug & Play” Domain. Methods Enzymol. 2018, 606, 1–71. [Google Scholar] [CrossRef]
- Wang, J.; Woldring, R.P.; Román-Meléndez, G.D.; McClain, A.M.; Alzua, B.R.; Marsh, E.N.G. Recent Advances in Radical SAM Enzymology: New Structures and Mechanisms. ACS Chem. Biol. 2014, 9, 1929–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booker, S.J.; Grove, T.L. Mechanistic and Functional Versatility of Radical SAM Enzymes. F1000 Biol. Rep. 2010, 2, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; van der Donk, W.A.; Liu, W. Radical-Mediated Enzymatic Methylation: A Tale of Two SAMS. Acc. Chem. Res. 2012, 45, 555–564. [Google Scholar] [CrossRef]
- Fujimori, D.G. Radical SAM-Mediated Methylation Reactions. Curr. Opin. Chem. Biol. 2013, 17, 597–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauerle, M.R.; Schwalm, E.L.; Booker, S.J. Mechanistic Diversity of Radical S-Adenosylmethionine (SAM)-Dependent Methylation. J. Biol. Chem. 2015, 290, 3995–4002. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Ribbe, M.W. Maturation of Nitrogenase Cofactor—The Role of a Class E Radical SAM Methyltransferase NifB. Curr. Opin. Chem. Biol. 2016, 31, 188–194. [Google Scholar] [CrossRef] [Green Version]
- Boal, A.K.; Grove, T.L.; McLaughlin, M.I.; Yennawar, N.H.; Booker, S.J.; Rosenzweig, A.C. Structural Basis for Methyl Transfer by a Radical SAM Enzyme. Science 2011, 332, 1089–1092. [Google Scholar] [CrossRef] [Green Version]
- Grove, T.L.; Benner, J.S.; Radle, M.I.; Ahlum, J.H.; Landgraf, B.J.; Krebs, C.; Booker, S.J. A Radically Different Mechanism for S-Adenosylmethionine–Dependent Methyltransferases. Science 2011, 332, 604–607. [Google Scholar] [CrossRef]
- McCusker, K.P.; Medzihradszky, K.F.; Shiver, A.L.; Nichols, R.J.; Yan, F.; Maltby, D.A.; Gross, C.A.; Galonić Fujimori, D. Covalent Intermediate in the Catalytic Mechanism of the Radical S-Adenosyl-l-Methionine Methyl Synthase RlmN Trapped by Mutagenesis. J. Am. Chem. Soc. 2012, 134, 18074–18081. [Google Scholar] [CrossRef] [Green Version]
- Silakov, A.; Grove, T.L.; Radle, M.I.; Bauerle, M.R.; Green, M.T.; Rosenzweig, A.C.; Boal, A.K.; Booker, S.J. Characterization of a Cross-Linked Protein–Nucleic Acid Substrate Radical in the Reaction Catalyzed by RlmN. J. Am. Chem. Soc. 2014, 136, 8221–8228. [Google Scholar] [CrossRef]
- Knox, H.L.; Chen, P.Y.-T.; Blaszczyk, A.J.; Mukherjee, A.; Grove, T.L.; Schwalm, E.L.; Wang, B.; Drennan, C.L.; Booker, S.J. Structural Basis for Non-Radical Catalysis by TsrM, a Radical-SAM Methylase. Nat. Chem. Biol. 2021, 17, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Knox, H.L.; Sinner, E.K.; Townsend, C.A.; Boal, A.K.; Booker, S.J. Structure of a B12-Dependent Radical SAM Enzyme in Carbapenem Biosynthesis. Nature 2022, 602, 343–348. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, W.-Q.; Zhu, X.; Zhang, Q. Functional Diversity of HemN-like Proteins. ACS Bio Med Chem Au 2022, 2, 109–119. [Google Scholar] [CrossRef]
- Layer, G.; Moser, J.; Heinz, D.W.; Jahn, D.; Schubert, W.-D. Crystal Structure of Coproporphyrinogen III Oxidase Reveals Cofactor Geometry of Radical SAM Enzymes. EMBO J. 2003, 22, 6214–6224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, C.T.; Iwig, D.F.; Wang, B.; Cossu, M.; Metcalf, W.W.; Boal, A.K.; Booker, S.J. Discovery, Structure and Mechanism of a Tetraether Lipid Synthase. Nature 2022, 609, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, E.L.; Grove, T.L.; Booker, S.J.; Boal, A.K. Crystallographic Capture of a Radical S-Adenosylmethionine Enzyme in the Act of Modifying TRNA. Science 2016, 352, 309–312. [Google Scholar] [CrossRef] [Green Version]
- Fajardo, A.S.; Legrand, P.; Payá-Tormo, L.A.; Martin, L.; Pellicer Martı Nez, M.T.; Echavarri-Erasun, C.; Vernède, X.; Rubio, L.M.; Nicolet, Y. Structural Insights into the Mechanism of the Radical SAM Carbide Synthase NifB, a Key Nitrogenase Cofactor Maturating Enzyme. J. Am. Chem. Soc. 2020, 142, 11006–11012. [Google Scholar] [CrossRef]
- Jenner, L.P.; Cherrier, M.V.; Amara, P.; Rubio, L.M.; Nicolet, Y. An Unexpected P-Cluster like Intermediate En Route to the Nitrogenase FeMo-Co. Chem. Sci. 2021, 12, 5269–5274. [Google Scholar] [CrossRef]
- Fyfe, C.D.; Bernardo-García, N.; Fradale, L.; Grimaldi, S.; Guillot, A.; Brewee, C.; Chavas, L.M.G.; Legrand, P.; Benjdia, A.; Berteau, O. Crystallographic Snapshots of a B12-Dependent Radical SAM Methyltransferase. Nature 2022, 602, 336–342. [Google Scholar] [CrossRef]
- Vázquez-Laslop, N.; Ramu, H.; Klepacki, D.; Kannan, K.; Mankin, A.S. The Key Function of a Conserved and Modified RRNA Residue in the Ribosomal Response to the Nascent Peptide. EMBO J. 2010, 29, 3108–3117. [Google Scholar] [CrossRef]
- Toh, S.-M.; Xiong, L.; Bae, T.; Mankin, A.S. The Methyltransferase YfgB/RlmN Is Responsible for Modification of Adenosine 2503 in 23S RRNA. RNA 2008, 14, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benítez-Páez, A.; Villarroya, M.; Armengod, M.-E. The Escherichia Coli RlmN Methyltransferase Is a Dual-Specificity Enzyme That Modifies Both RRNA and TRNA and Controls Translational Accuracy. RNA 2012, 18, 1783–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giessing, A.M.B.; Jensen, S.S.; Rasmussen, A.; Hansen, L.H.; Gondela, A.; Long, K.; Vester, B.; Kirpekar, F. Identification of 8-Methyladenosine as the Modification Catalyzed by the Radical SAM Methyltransferase Cfr That Confers Antibiotic Resistance in Bacteria. RNA 2009, 15, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehrenberg, C.; Schwarz, S.; Jacobsen, L.; Hansen, L.H.; Vester, B. A New Mechanism for Chloramphenicol, Florfenicol and Clindamycin Resistance: Methylation of 23S Ribosomal RNA at A2503. Mol. Microbiol. 2005, 57, 1064–1073. [Google Scholar] [CrossRef]
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. The Cfr RRNA Methyltransferase Confers Resistance to Phenicols, Lincosamides, Oxazolidinones, Pleuromutilins, and Streptogramin A Antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2500–2505. [Google Scholar] [CrossRef] [Green Version]
- Hioe, J.; Zipse, H. Hydrogen Transfer in SAM-Mediated Enzymatic Radical Reactions. Chem. Eur. J. 2012, 18, 16463–16472. [Google Scholar] [CrossRef]
- Grove, T.L.; Livada, J.; Schwalm, E.L.; Green, M.T.; Booker, S.J.; Silakov, A. A Substrate Radical Intermediate in Catalysis by the Antibiotic Resistance Protein Cfr. Nat. Chem. Biol. 2013, 9, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Minisci, F.; Bernardi, R.; Bertini, F.; Galli, R.; Perchinummo, M. Nucleophilic Character of Alkyl Radicals—VI: A New Convenient Selective Alkylation of Heteroaromatic Bases. Tetrahedron 1971, 27, 3575–3579. [Google Scholar] [CrossRef]
- Challand, M.R.; Salvadori, E.; Driesener, R.C.; Kay, C.W.M.; Roach, P.L.; Spencer, J. Cysteine Methylation Controls Radical Generation in the Cfr Radical AdoMet RRNA Methyltransferase. PLoS ONE 2013, 8, e67979. [Google Scholar] [CrossRef] [Green Version]
- Kaminska, K.H.; Purta, E.; Hansen, L.H.; Bujnicki, J.M.; Vester, B.; Long, K.S. Insights into the Structure, Function and Evolution of the Radical-SAM 23S RRNA Methyltransferase Cfr That Confers Antibiotic Resistance in Bacteria. Nucleic Acids Res. 2010, 38, 1652–1663. [Google Scholar] [CrossRef]
- Hutcheson, R.U.; Broderick, J.B. Radical SAM Enzymes in Methylation and Methylthiolation. Met. Integr. Biometal Sci. 2012, 4, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Dong, L.; Liu, Y. A QM/MM Study of the Catalytic Mechanism of SAM Methyltransferase RlmN from Escherichia Coli. Proteins 2017, 85, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Ntokou, E.; Hansen, L.H.; Kongsted, J.; Vester, B. Biochemical and Computational Analysis of the Substrate Specificities of Cfr and RlmN Methyltransferases. PLoS ONE 2015, 10, e0145655. [Google Scholar] [CrossRef] [Green Version]
- Berteau, O.; Benjdia, A. DNA Repair by the Radical SAM Enzyme Spore Photoproduct Lyase: From Biochemistry to Structural Investigations. Photochem. Photobiol. 2017, 93, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandor-Proust, A.; Berteau, O.; Douki, T.; Gasparutto, D.; Ollagnier-de-Choudens, S.; Fontecave, M.; Atta, M. DNA Repair and Free Radicals, New Insights into the Mechanism of Spore Photoproduct Lyase Revealed by Single Amino Acid Substitution. J. Biol. Chem. 2008, 283, 36361–36368. [Google Scholar] [CrossRef] [Green Version]
- Benjdia, A.; Heil, K.; Winkler, A.; Carell, T.; Schlichting, I. Rescuing DNA Repair Activity by Rewiring the H-Atom Transfer Pathway in the Radical SAM Enzyme, Spore Photoproduct Lyase. Chem. Commun. Camb. Engl. 2014, 50, 14201–14204. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.C. Cobalamin-Dependent Radical S-Adenosyl-L-Methionine Enzymes in Natural Product Biosynthesis. Nat. Prod. Rep. 2018, 35, 707–720. [Google Scholar] [CrossRef]
- Pierre, S.; Guillot, A.; Benjdia, A.; Sandström, C.; Langella, P.; Berteau, O. Thiostrepton Tryptophan Methyltransferase Expands the Chemistry of Radical SAM Enzymes. Nat. Chem. Biol. 2012, 8, 957–959. [Google Scholar] [CrossRef]
- Li, C.; Kelly, W.L. Recent Advances in Thiopeptide Antibiotic Biosynthesis. Nat. Prod. Rep. 2010, 27, 153–164. [Google Scholar] [CrossRef]
- Wu, R.; Ding, W.; Zhang, Q. Consecutive Methylation Catalyzed by TsrM, an Atypical Class B Radical SAM Methylase. Chin. J. Chem. 2022, 40, 1693–1698. [Google Scholar] [CrossRef]
- Soualmia, F.; Guillot, A.; Sabat, N.; Brewee, C.; Kubiak, X.; Haumann, M.; Guinchard, X.; Benjdia, A.; Berteau, O. Exploring the Biosynthetic Potential of TsrM, a B12-Dependent Radical SAM Methyltransferase Catalyzing Non-Radical Reactions. Chem. Eur. J. 2022, 28, e202200627. [Google Scholar] [CrossRef] [PubMed]
- Sinner, E.K.; Lichstrahl, M.S.; Li, R.; Marous, D.R.; Townsend, C.A. Methylations in Complex Carbapenem Biosynthesis Are Catalyzed by a Single Cobalamin-Dependent Radical S-Adenosylmethionine Enzyme. Chem. Commun. Camb. Engl. 2019, 55, 14934–14937. [Google Scholar] [CrossRef]
- Sinner, E.K.; Marous, D.R.; Townsend, C.A. Evolution of Methods for the Study of Cobalamin-Dependent Radical SAM Enzymes. ACS Bio Med Chem Au 2022, 2, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Deobald, D.; Adrian, L.; Schöne, C.; Rother, M.; Layer, G. Identification of a Unique Radical SAM Methyltransferase Required for the Sp3-C-Methylation of an Arginine Residue of Methyl-Coenzyme M Reductase. Sci. Rep. 2018, 8, 7404. [Google Scholar] [CrossRef] [PubMed]
- Radle, M.I.; Miller, D.V.; Laremore, T.N.; Booker, S.J. Methanogenesis Marker Protein 10 (Mmp10) from Methanosarcina Acetivorans Is a Radical S-Adenosylmethionine Methylase That Unexpectedly Requires Cobalamin. J. Biol. Chem. 2019, 294, 11712–11725. [Google Scholar] [CrossRef] [Green Version]
- Lyu, Z.; Shao, N.; Chou, C.-W.; Shi, H.; Patel, R.; Duin, E.C.; Whitman, W.B. Posttranslational Methylation of Arginine in Methyl Coenzyme M Reductase Has a Profound Impact on Both Methanogenesis and Growth of Methanococcus Maripaludis. J. Bacteriol. 2020, 202, e00654-19. [Google Scholar] [CrossRef]
- Jarrett, J.T. Surprise! A Hidden B12 Cofactor Catalyzes a Radical Methylation. J. Biol. Chem. 2019, 294, 11726–11727. [Google Scholar] [CrossRef] [Green Version]
- Blaszczyk, A.J.; Silakov, A.; Zhang, B.; Maiocco, S.J.; Lanz, N.D.; Kelly, W.L.; Elliott, S.J.; Krebs, C.; Booker, S.J. Spectroscopic and Electrochemical Characterization of the Iron–Sulfur and Cobalamin Cofactors of TsrM, an Unusual Radical S-Adenosylmethionine Methylase. J. Am. Chem. Soc. 2016, 138, 3416–3426. [Google Scholar] [CrossRef]
- Blaszczyk, A.J.; Wang, B.; Silakov, A.; Ho, J.V.; Booker, S.J. Efficient Methylation of C2 in L-Tryptophan by the Cobalamin-Dependent Radical S-Adenosylmethionine Methylase TsrM Requires an Unmodified N1 Amine. J. Biol. Chem. 2017, 292, 15456–15467. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, E.C.; Drennan, C.L. The Atypical Cobalamin-Dependent S-Adenosyl-l-Methionine Nonradical Methylase TsrM and Its Radical Counterparts. J. Am. Chem. Soc. 2022, 144, 5673–5684. [Google Scholar] [CrossRef]
- Moore, B.N.; Julian, R.R. Dissociation Energies of X–H Bonds in Amino Acids. Phys. Chem. Chem. Phys. 2012, 14, 3148–3154. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W.M. CRC Handbook of Chemistry and Physics, 96th ed.; CRC Press: Boca Raton, FL, USA, 2015; ISBN 978-1-4822-6097-7. [Google Scholar]
- Bridwell-Rabb, J.; Li, B.; Drennan, C.L. Cobalamin-Dependent Radical S-Adenosylmethionine Enzymes: Capitalizing on Old Motifs for New Functions. ACS Bio Med Chem Au 2022, 2, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.-H.; Liao, R.-Z. Computational Study Revealed a “Pull–Push” Radical Transfer Mechanism of Mmp10-Catalyzed Cδ-Methylation of Arginine. Chem. Commun. 2022, 58, 7144–7147. [Google Scholar] [CrossRef] [PubMed]
- Lichstrahl, M.S.; Townsend, C.A.; Sinner, E.K. Stereochemical Course of Cobalamin-Dependent Radical SAM Methylation by TokK and ThnK. RSC Chem. Biol. 2022, 3, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, A.J.; Knox, H.L.; Booker, S.J. Understanding the Role of Electron Donors in the Reaction Catalyzed by TsrM, a Cobalamin-Dependent Radical S-Adenosylmethionine Methylase. J. Biol. Inorg. Chem. JBIC 2019, 24, 831–839. [Google Scholar] [CrossRef]
- Goldman, P.J.; Grove, T.L.; Booker, S.J.; Drennan, C.L. X-Ray Analysis of Butirosin Biosynthetic Enzyme BtrN Redefines Structural Motifs for AdoMet Radical Chemistry. Proc. Natl. Acad. Sci. USA 2013, 110, 15949–15954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, D.P.; Vey, J.L.; Croft, A.K.; Drennan, C.L. Structural Diversity in the AdoMet Radical Enzyme Superfamily. Biochim. Biophys. Acta 2012, 1824, 1178–1195. [Google Scholar] [CrossRef] [Green Version]
- Fay, A.W.; Wiig, J.A.; Lee, C.C.; Hu, Y. Identification and Characterization of Functional Homologs of Nitrogenase Cofactor Biosynthesis Protein NifB from Methanogens. Proc. Natl. Acad. Sci. USA 2015, 112, 14829–14833. [Google Scholar] [CrossRef] [Green Version]
- Curatti, L.; Ludden, P.W.; Rubio, L.M. NifB-Dependent in Vitro Synthesis of the Iron-Molybdenum Cofactor of Nitrogenase. Proc. Natl. Acad. Sci. USA 2006, 103, 5297–5301. [Google Scholar] [CrossRef] [Green Version]
- Burgess, B.K.; Lowe, D.J. Mechanism of Molybdenum Nitrogenase. Chem. Rev. 1996, 96, 2983–3012. [Google Scholar] [CrossRef]
- Curatti, L.; Hernandez, J.A.; Igarashi, R.Y.; Soboh, B.; Zhao, D.; Rubio, L.M. In Vitro Synthesis of the Iron-Molybdenum Cofactor of Nitrogenase from Iron, Sulfur, Molybdenum, and Homocitrate Using Purified Proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 17626–17631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, S.L.; Bakovic, S.I.P.; Duda, R.D.; Maheshwari, S.; Milton, R.D.; Minteer, S.D.; Janik, M.J.; Renner, J.N.; Greenlee, L.F. Catalysts for Nitrogen Reduction to Ammonia. Nat. Catal. 2018, 1, 490–500. [Google Scholar] [CrossRef]
- Lancaster, K.M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.; Ribbe, M.W.; Neese, F.; Bergmann, U.; DeBeer, S. X-Ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spatzal, T.; Aksoyoglu, M.; Zhang, L.; Andrade, S.L.A.; Schleicher, E.; Weber, S.; Rees, D.C.; Einsle, O. Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940. [Google Scholar] [CrossRef] [Green Version]
- Wiig, J.A.; Hu, Y.; Chung Lee, C.; Ribbe, M.W. Radical SAM-Dependent Carbon Insertion into the Nitrogenase M-Cluster. Science 2012, 337, 1672–1675. [Google Scholar] [CrossRef] [Green Version]
- Burén, S.; Jiménez-Vicente, E.; Echavarri-Erasun, C.; Rubio, L.M. Biosynthesis of Nitrogenase Cofactors. Chem. Rev. 2020, 120, 4921–4968. [Google Scholar] [CrossRef] [Green Version]
- Wilcoxen, J.; Arragain, S.; Scandurra, A.A.; Jimenez-Vicente, E.; Echavarri-Erasun, C.; Pollmann, S.; Britt, R.D.; Rubio, L.M. Electron Paramagnetic Resonance Characterization of Three Iron–Sulfur Clusters Present in the Nitrogenase Cofactor Maturase NifB from Methanocaldococcus Infernus. J. Am. Chem. Soc. 2016, 138, 7468–7471. [Google Scholar] [CrossRef] [Green Version]
- Rettberg, L.A.; Wilcoxen, J.; Lee, C.C.; Stiebritz, M.T.; Tanifuji, K.; Britt, R.D.; Hu, Y. Probing the Coordination and Function of Fe4S4 Modules in Nitrogenase Assembly Protein NifB. Nat. Commun. 2018, 9, 2824. [Google Scholar] [CrossRef] [Green Version]
- Wiig, J.A.; Hu, Y.; Ribbe, M.W. Refining the Pathway of Carbide Insertion into the Nitrogenase M-Cluster. Nat. Commun. 2015, 6, 8034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, W.; Rettberg, L.; Stiebritz, M.; Jasniewski, A.; Tanifuji, K.; Lee, C.; Ribbe, M.; Hu, Y. Crystallographic Analysis of NifB with a Full Complement of Clusters: Structural Insights into the Radical SAM-Dependent Carbide Insertion during Nitrogenase Cofactor Assembly. Angew. Chem. Int. Ed. Engl. 2020, 60, 2364–2370. [Google Scholar] [CrossRef]
- Nicolet, Y.; Cherrier, M.V.; Amara, P. Radical SAM Enzymes and Metallocofactor Assembly: A Structural Point of View. ACS Bio Med Chem Au 2022, 2, 36–52. [Google Scholar] [CrossRef]
- Jiménez-Vicente, E.; Navarro-Rodríguez, M.; Poza-Carrión, C.; Rubio, L.M. Role of Azotobacter Vinelandii FdxN in FeMo-Co Biosynthesis. FEBS Lett. 2014, 588, 512–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rettberg, L.A.; Wilcoxen, J.; Jasniewski, A.J.; Lee, C.C.; Tanifuji, K.; Hu, Y.; Britt, R.D.; Ribbe, M.W. Identity and Function of an Essential Nitrogen Ligand of the Nitrogenase Cofactor Biosynthesis Protein NifB. Nat. Commun. 2020, 11, 1757. [Google Scholar] [CrossRef] [Green Version]
- Tanifuji, K.; Lee, C.C.; Sickerman, N.S.; Tatsumi, K.; Ohki, Y.; Hu, Y.; Ribbe, M.W. Tracing the “ninth Sulfur” of the Nitrogenase Cofactor via a Semi-Synthetic Approach. Nat. Chem. 2018, 10, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Jasniewski, A.J.; Wilcoxen, J.; Tanifuji, K.; Hedman, B.; Hodgson, K.O.; Britt, R.D.; Hu, Y.; Ribbe, M.W. Spectroscopic Characterization of an Eight-Iron Nitrogenase Cofactor Precursor That Lacks the “9th Sulfur”. Angew. Chem. Int. Ed. Engl. 2019, 58, 14703–14707. [Google Scholar] [CrossRef]
- Tanifuji, K.; Jasniewski, A.J.; Villarreal, D.; Stiebritz, M.T.; Lee, C.C.; Wilcoxen, J.; Okhi, Y.; Chatterjee, R.; Bogacz, I.; Yano, J.; et al. Tracing the Incorporation of the “Ninth Sulfur” into the Nitrogenase Cofactor Precursor with Selenite and Tellurite. Nat. Chem. 2021, 13, 1228–1234. [Google Scholar] [CrossRef]
- Kessler, D. Enzymatic Activation of Sulfur for Incorporation into Biomolecules in Prokaryotes. FEMS Microbiol. Rev. 2006, 30, 825–840. [Google Scholar] [CrossRef] [Green Version]
- Arragain, S.; Jiménez-Vicente, E.; Scandurra, A.A.; Burén, S.; Rubio, L.M.; Echavarri-Erasun, C. Diversity and Functional Analysis of the FeMo-Cofactor Maturase NifB. Front. Plant Sci. 2017, 8, 1947. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, J.A.; Igarashi, R.Y.; Soboh, B.; Curatti, L.; Dean, D.R.; Ludden, P.W.; Rubio, L.M. NifX and NifEN Exchange NifB Cofactor and the VK-Cluster, a Newly Isolated Intermediate of the Iron-Molybdenum Cofactor Biosynthetic Pathway. Mol. Microbiol. 2007, 63, 177–192. [Google Scholar] [CrossRef]
- Rangaraj, P.; Ruttimann-Johnson, C.; Shah, V.K.; Ludden, P.W. Accumulation of 55Fe-Labeled Precursors of the Iron-Molybdenum Cofactor of Nitrogenase on NifH and NifX of Azotobacter Vinelandii. J. Biol. Chem. 2001, 276, 15968–15974. [Google Scholar] [CrossRef]
- Burén, S.; Jiang, X.; López-Torrejón, G.; Echavarri-Erasun, C.; Rubio, L.M. Purification and In Vitro Activity of Mitochondria Targeted Nitrogenase Cofactor Maturase NifB. Front. Plant Sci. 2017, 8, 1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.; Li, Y.; Zhao, J.; Ji, X.; Mo, T.; Qianzhu, H.; Tu, T.; Deng, Z.; Yu, Y.; Chen, F.; et al. The Catalytic Mechanism of the Class C Radical S-Adenosylmethionine Methyltransferase NosN. Angew. Chem. Int. Ed. 2017, 56, 3857–3861. [Google Scholar] [CrossRef] [PubMed]
- LaMattina, J.W.; Wang, B.; Badding, E.D.; Gadsby, L.K.; Grove, T.L.; Booker, S.J. The Radical S-Adenosylmethionine Methylase NosN Catalyzes Both C1 Transfer and Formation of the Ester Linkage of the Side-Ring System during the Biosynthesis of Nosiheptide. J. Am. Chem. Soc. 2017, 139, 17438–17445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.; Mandalapu, D.; Cheng, J.; Ding, W.; Zhang, Q. Expanding the Chemistry of the Class C Radical SAM Methyltransferase NosN by Using an Allyl Analogue of SAM. Angew. Chem. Int. Ed. 2018, 57, 6601–6604. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Liu, Q.; Liu, X.-M.; Shi, H.-W.; Chen, S.-F. Using Synthetic Biology to Increase Nitrogenase Activity. Microb. Cell Factories 2016, 15, 43. [Google Scholar] [CrossRef] [Green Version]
- Gui, J.; Zhou, Q.; Pan, C.-M.; Yabe, Y.; Burns, A.C.; Collins, M.R.; Ornelas, M.A.; Ishihara, Y.; Baran, P.S. C–H Methylation of Heteroarenes Inspired by Radical SAM Methyl Transferase. J. Am. Chem. Soc. 2014, 136, 4853–4856. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.-Q.; Nicolet, Y. Structure and Catalytic Mechanism of Radical SAM Methylases. Life 2022, 12, 1732. https://doi.org/10.3390/life12111732
Nguyen T-Q, Nicolet Y. Structure and Catalytic Mechanism of Radical SAM Methylases. Life. 2022; 12(11):1732. https://doi.org/10.3390/life12111732
Chicago/Turabian StyleNguyen, Tu-Quynh, and Yvain Nicolet. 2022. "Structure and Catalytic Mechanism of Radical SAM Methylases" Life 12, no. 11: 1732. https://doi.org/10.3390/life12111732
APA StyleNguyen, T. -Q., & Nicolet, Y. (2022). Structure and Catalytic Mechanism of Radical SAM Methylases. Life, 12(11), 1732. https://doi.org/10.3390/life12111732