
2-(3-Bromophenyl)-8-fluoroquinazoline-4-carboxylic Acid as a Novel and Selective Aurora A Kinase Inhibitory Lead with Apoptosis Properties: Design, Synthesis, In Vitro and In Silico Biological Evaluation

 , ,
, ,  , , , ,
, , , , 
Abstract
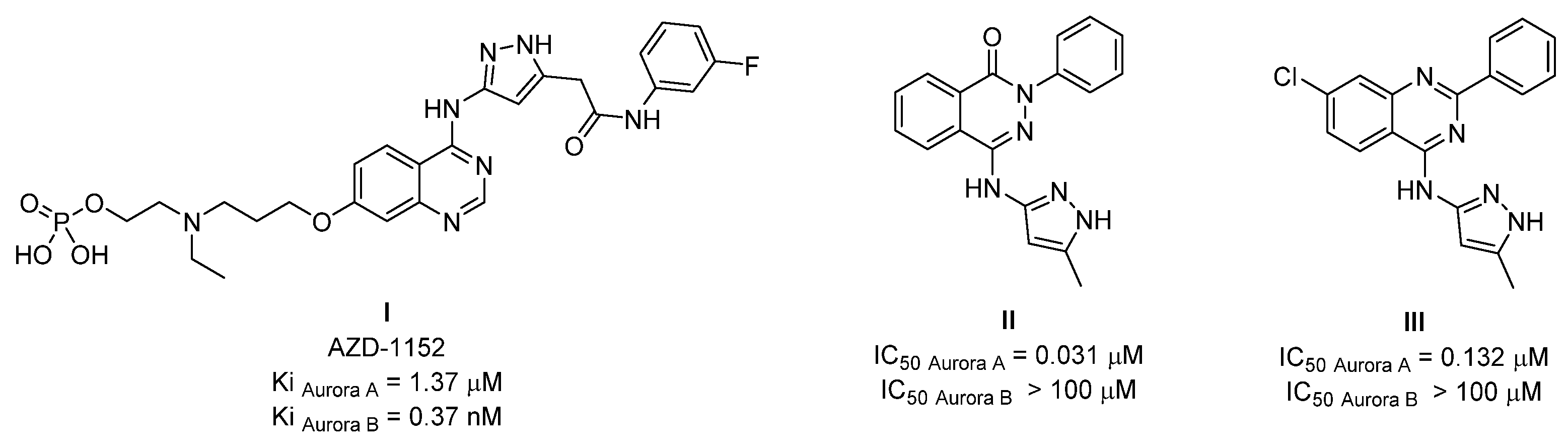
:1. Introduction
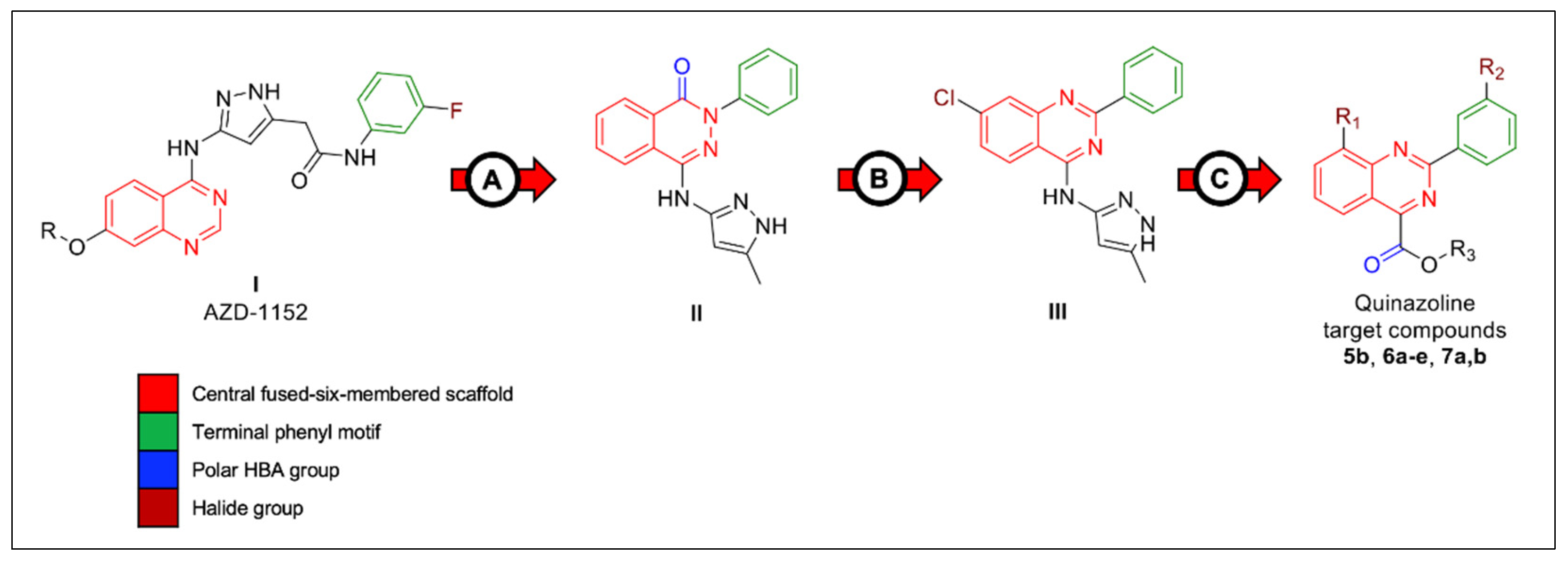
2. Results and Discussion
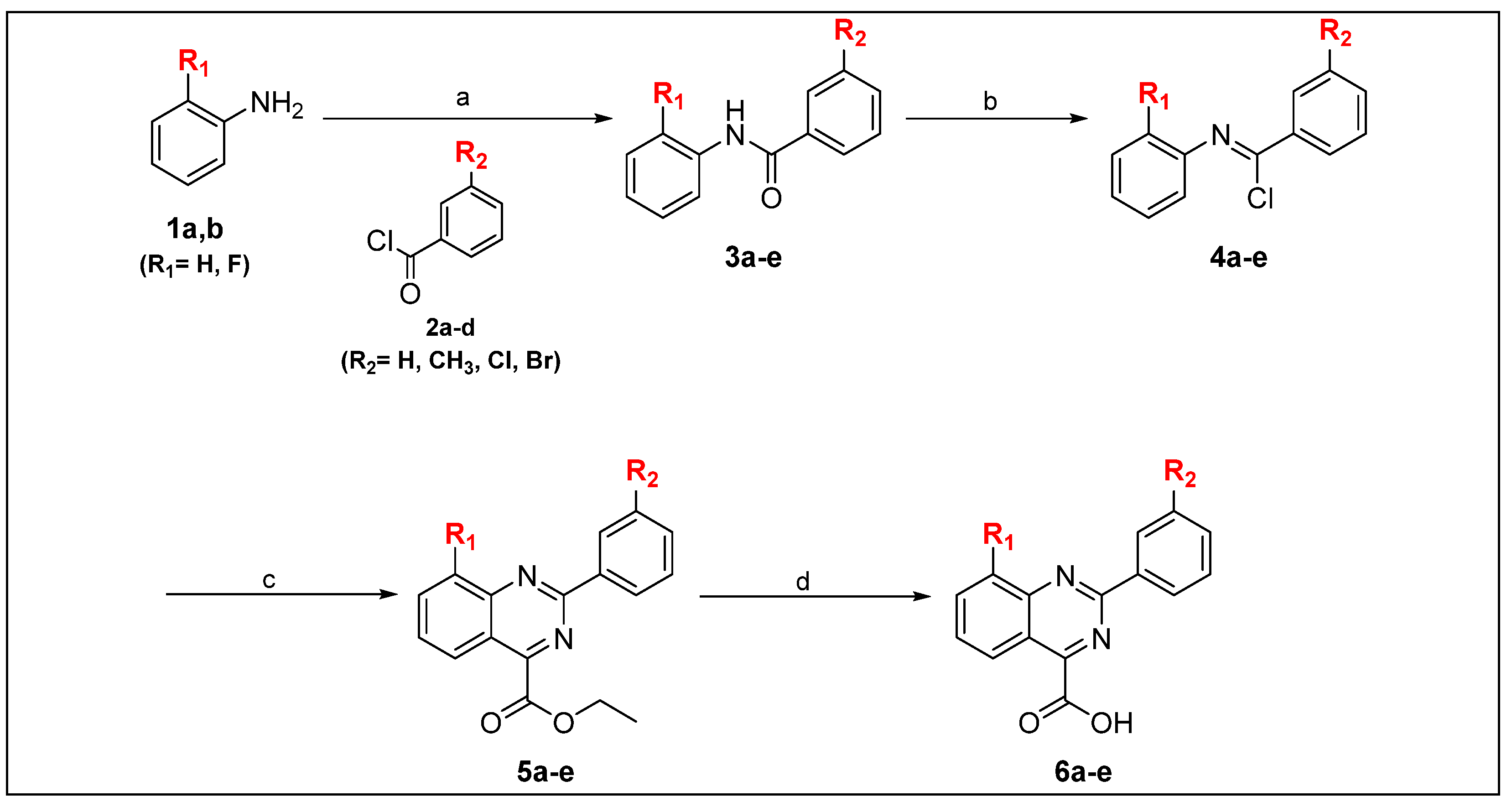
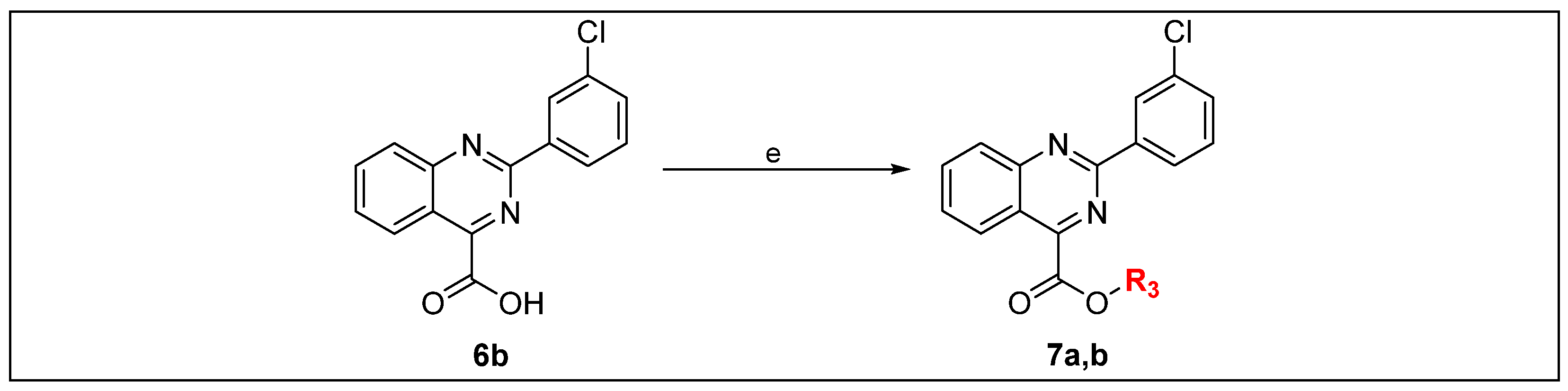
2.1. Chemistry
2.2. In Vitro Screening
2.2.1. Kinase Inhibitory Assay
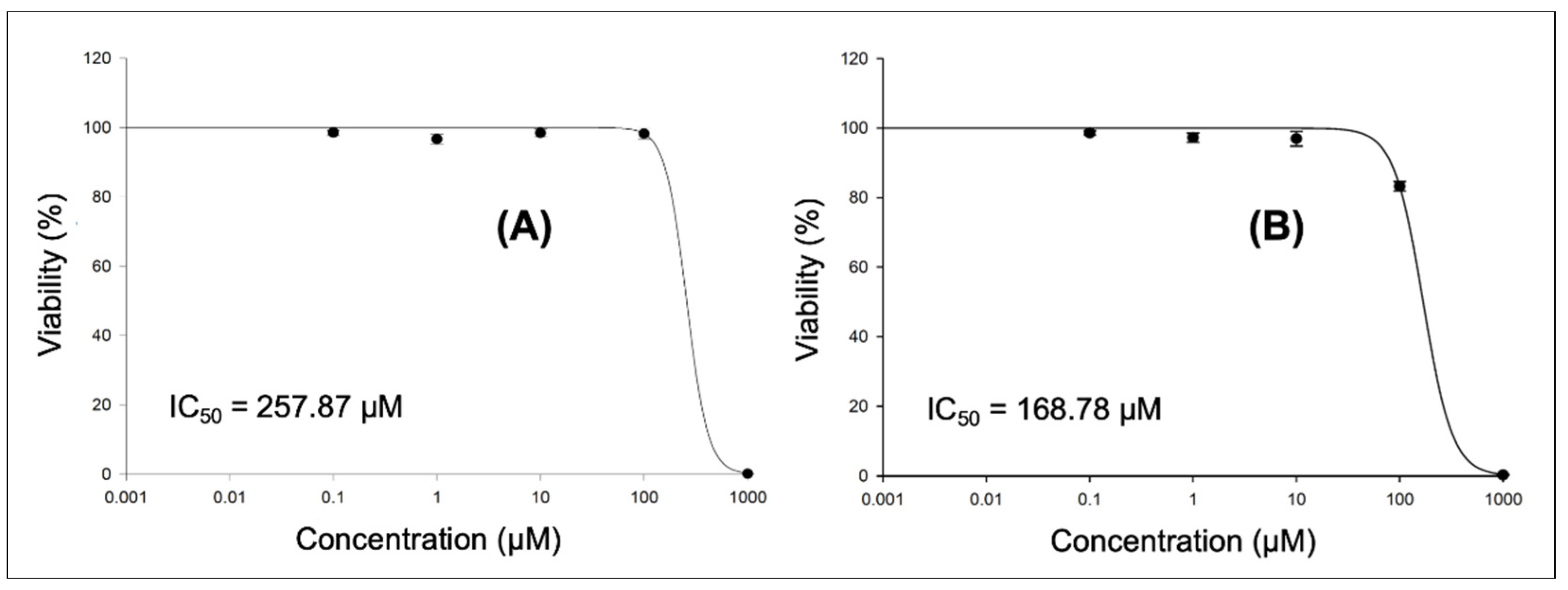
2.2.2. Cytotoxicity Assay
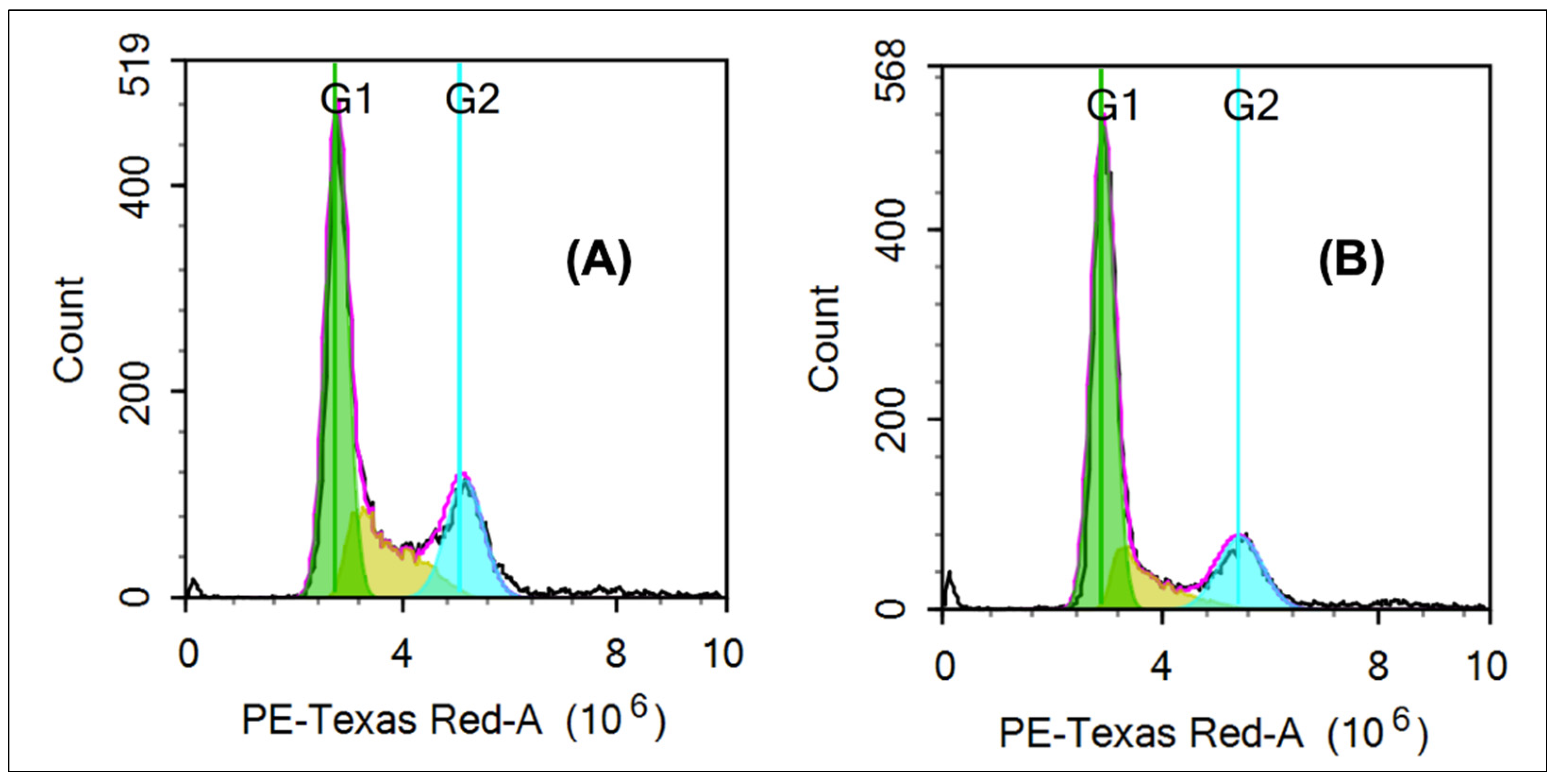
2.2.3. Cell Cycle Assay
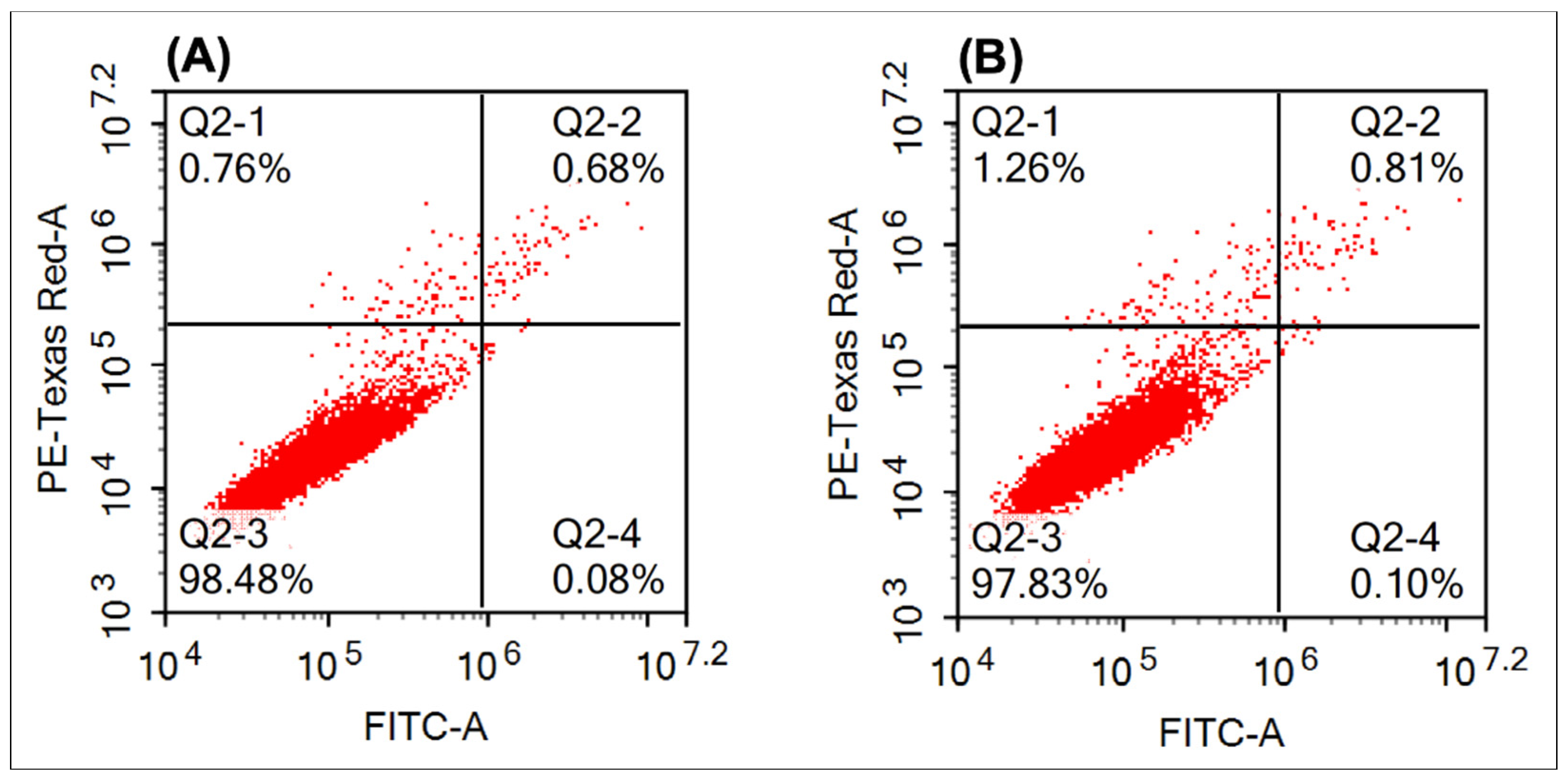
2.2.4. Apoptosis
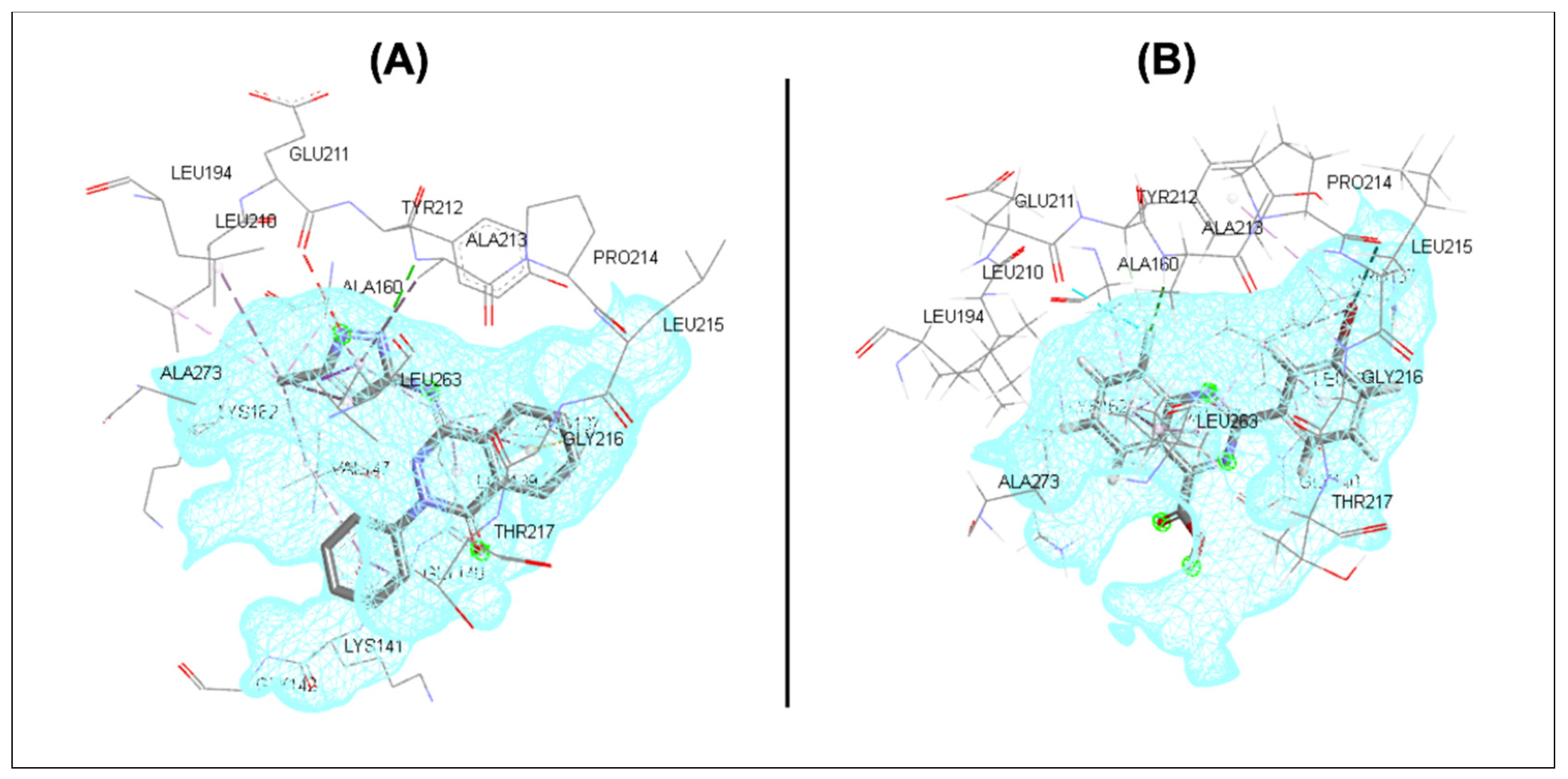
2.3. In Silico Screening
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. General Procedure for the Synthesis of Derivatives 3a–e
3.1.3. General Procedure for the Synthesis of Derivatives 4a–e
3.1.4. General Procedure for the Synthesis of Derivatives 5a–e
3.1.5. General Procedure for the Synthesis of Derivatives 6a–e
3.1.6. General Procedure for the Synthesis of Derivatives 7a,b
3.2. In Vitro Screening
3.2.1. Kinase Inhibitory Assay
3.2.2. Cytotoxicity Assay
3.2.3. Cell Cycle
3.2.4. Apoptosis
3.3. In Silico Screening
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Borisa, A.C.; Bhatt, H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017, 140, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.D. Aurora kinases: Shining lights on the therapeutic horizon? Oncogene 2005, 24, 5005–5015. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Wang, Y.-H.; Zhuo, J.-X.; Tu, Z.-C.; Wu, R.; Yan, M.; Liu, Q.; Lu, G. Structure-based drug design: Synthesis and biological evaluation of quinazolin-4-amine derivatives as selective Aurora A kinase inhibitors. Eur. J. Med. Chem. 2018, 157, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, T.; Gupta, O.; Singh, G.; Monga, V. Aurora kinase inhibitors as potential anticancer agents: Recent advances. Eur. J. Med. Chem. 2021, 221, 113495. [Google Scholar] [CrossRef]
- Fu, S.; Li, Y.; Huang, J.; Liu, T.; Hong, Z.; Chen, A.; Bast, R.C.; Kavanagh, J.J.; Gershenson, D.M.; Sood, A.K. Aurora kinase inhibitor VE 465 synergistically enhances cytotoxicity of carboplatin in ovarian cancer cells through induction of apoptosis and downregulation of histone 3. Cancer Biol. Ther. 2012, 13, 1034–1041. [Google Scholar] [CrossRef]
- Lindon, C.; Grant, R.; Min, M. Ubiquitin-mediated degradation of aurora kinases. Front. Oncol. 2016, 5, 307. [Google Scholar] [CrossRef]
- Fan, C.; Zhong, T.; Yang, H.; Yang, Y.; Wang, D.; Yang, X.; Xu, Y.; Fan, Y. Design, synthesis, biological evaluation of 6-(2-amino-1H-benzo [d] imidazole-6-yl) quinazolin-4 (3H)-one derivatives as novel anticancer agents with Aurora kinase inhibition. Eur. J. Med. Chem. 2020, 190, 112108. [Google Scholar] [CrossRef]
- Bavetsias, V.; Faisal, A.; Crumpler, S.; Brown, N.; Kosmopoulou, M.; Joshi, A.; Atrash, B.; Pérez-Fuertes, Y.; Schmitt, J.A.; Boxall, K.J. Aurora isoform selectivity: Design and synthesis of imidazo [4,5-b] pyridine derivatives as highly selective inhibitors of Aurora-A kinase in cells. J. Med. Chem. 2013, 56, 9122–9135. [Google Scholar] [CrossRef]
- Pollard, J.R.; Mortimore, M. Discovery and development of aurora kinase inhibitors as anticancer agents. J. Med. Chem. 2009, 52, 2629–2651. [Google Scholar] [CrossRef]
- Green, M.R.; Woolery, J.E.; Mahadevan, D. Update on aurora kinase targeted therapeutics in oncology. Expert Opin. Drug Dis. 2011, 6, 291–307. [Google Scholar] [CrossRef]
- Cheung, C.H.A.; Coumar, M.S.; Chang, J.Y.; Hsieh, H.P. Aurora kinase inhibitor patents and agents in clinical testing: An update (2009–10). Expert Opin. Ther. Pat. 2011, 21, 857–884. [Google Scholar] [CrossRef] [PubMed]
- Mortlock, A.A.; Foote, K.M.; Heron, N.M.; Jung, F.H.; Pasquet, G.; Lohmann, J.-J.M.; Warin, N.; Renaud, F.; De Savi, C.; Roberts, N.J. Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J. Med. Chem. 2007, 50, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Aliagas-Martin, I.; Burdick, D.; Corson, L.; Dotson, J.; Drummond, J.; Fields, C.; Huang, O.W.; Hunsaker, T.; Kleinheinz, T.; Krueger, E.; et al. A class of 2,4-bisanilinopyrimidine Aurora A inhibitors with unusually high selectivity against Aurora B. J. Med. Chem. 2009, 52, 3300–3307. [Google Scholar] [CrossRef] [PubMed]
- White, T.D.; Berglund, K.D.; Groh, J.M.; Johnson, M.D.; Miller, R.D.; Yates, M.H. Development of a continuous Schotten–Baumann route to an acyl sulfonamide. Org. Process Res. Dev. 2012, 16, 939–957. [Google Scholar] [CrossRef]
- Prime, M.E.; Courtney, S.M.; Brookfield, F.A.; Marston, R.W.; Walker, V.; Warne, J.; Boyd, A.E.; Kairies, N.A.; von der Saal, W.; Limberg, A.; et al. Phthalazinone pyrazoles as potent, selective, and orally bioavailable inhibitors of Aurora-A kinase. J. Med. Chem. 2011, 54, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-E.; Elkamhawy, A.; Hassan, A.H.; Pae, A.N.; Lee, J.; Paik, S.; Park, B.-G.; Roh, E.J. Synthesis and evaluation of new pyridyl/pyrazinyl thiourea derivatives: Neuroprotection against amyloid-β-induced toxicity. Eur. J. Med. Chem. 2017, 141, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Shi, X.; He, G.; Tong, Y.; Jiang, Z.; Han, S. One-pot synthesis of 2-arylbenzoxazole derivatives via Cu (I) catalyzed C–N/C–O coupling of N-(2-chloro-phenyl)-2-halo-benzamides and primary amines. Tetrahedron 2015, 71, 431–435. [Google Scholar] [CrossRef]
- Zhao, H.-B.; Hou, Z.-W.; Liu, Z.-J.; Zhou, Z.-F.; Song, J.; Xu, H.-C. Amidinyl radical formation through anodic N−H bond cleavage and its application in aromatic C−H bond functionalization. Angew. Chem. Int. Ed. 2017, 56, 587–590. [Google Scholar] [CrossRef]
- Giardina, G.A.M.; Sarau, H.M.; Farina, C.; Medhurst, A.D.; Grugni, M.; Raveglia, L.F.; Schmidt, D.B.; Rigolio, R.; Luttmann, M.; Vecchietti, V.; et al. Discovery of a novel class of selective non-peptide antagonists for the human neurokinin-3 receptor. 1. Identification of the 4-quinolinecarboxamide framework. J. Med. Chem. 1997, 40, 1794–1807. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Ammar, U.M.; Paik, S.; Abdellattif, M.H.; Elsherbeny, M.H.; Lee, K.; Roh, E.J. Scaffold repurposing of in-house small molecule candidates leads to discovery of first-in-class CDK-1/HER-2 dual inhibitors: In vitro and in silico screening. Molecules 2021, 26, 5324. [Google Scholar] [CrossRef]
- Lee, K.; Nada, H.; Byun, H.J.; Lee, C.H.; Elkamhawy, A. Hit Identification of a Novel Quinazoline Sulfonamide as a Promising EphB3 Inhibitor: Design, Virtual Combinatorial Library, Synthesis, Biological Evaluation, and Docking Simulation Studies. Pharmaceuticals 2021, 14, 1247. [Google Scholar] [CrossRef] [PubMed]
- Al-Sanea, M.M.; Elkamhawy, A.; Zakaria, A.; Park, B.S.; Kwon, Y.; Lee, S.H.; Lee, S.W.; Kim, I.T. Synthesis and in vitro screening of phenylbipyridinylpyrazole derivatives as potential antiproliferative agents. Molecules 2015, 20, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Allam, R.M.; Al-Abd, A.M.; Khedr, A.; Sharaf, O.A.; Nofal, S.M.; Khalifa, A.E.; Mosli, H.A.; Abdel-Naim, A.B. Fingolimod interrupts the cross talk between estrogen metabolism and sphingolipid metabolism within prostate cancer cells. Toxicol. Lett. 2018, 291, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Fekry, M.; Ezzat, S.M.; Salama, M.M.; AlShehri, O.Y.; Al-Abd, A.M. Bioactive glycoalkaloides isolated from Solanum melongena fruit peels with potential anticancer properties against hepatocellular carcinoma cells. Sci. Rep. 2019, 9, 1746. [Google Scholar] [CrossRef]
- Baghdadi, M.A.; Al-Abbasi, F.A.; El-Halawany, A.M.; Aseeri, A.H.; Al-Abd, A.M. Anticancer profiling for coumarins and related O-naphthoquinones from mansonia gagei against solid tumor cells in vitro. Molecules 2018, 23, 1020. [Google Scholar] [CrossRef]
- Alaufi, O.M.; Noorwali, A.; Zahran, F.; Al-Abd, A.M.; Al-Attas, S. Cytotoxicity of thymoquinone alone or in combination with cisplatin (CDDP) against oral squamous cell carcinoma in vitro. Sci. Rep. 2017, 7, 13131. [Google Scholar] [CrossRef]
- Mohamed, G.A.; Al-Abd, A.M.; El-Halawany, A.M.; Abdallah, H.; Ibrahim, S.R.M. New xanthones and cytotoxic constituents from Garcinia mangostana fruit hulls against human hepatocellular, breast, and colorectal cancer cell lines. J. Enthnopharmacol. 2017, 198, 302–312. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | R2 | R3 |
|---|---|---|---|
| 5b | H | Cl | Et |
| 6a | H | H | H |
| 6b | H | Cl | H |
| 6c | F | Me | H |
| 6d | F | H | H |
| 6e | F | Br | H |
| 7a | H | Cl | Me |
| 7b | H | Cl | i-Pr |
| Compound | Aurora A (Inhibition%) |
|---|---|
| 6a | 7.64 ± 1.19 |
| 6b | 28.48 ± 2.40 |
| 6c | 14.83 ± 0.14 |
| 6d | 22.18 ± 1.86 |
| 6e | 51.78 ± 0.38 |
| Compound | Aurora A (Inhibition%) |
|---|---|
| 5b | 6.53 ± 0.58 |
| 7a | −2.61 ± 0.15 |
| 7b | 3.83 ± 1.38 |
| Kinase | Compound 6e (Activity%) | Kinase | Compound 6e (Activity%) |
|---|---|---|---|
| ABL-1 | 106.77 | CDK-2/cyclin A | 83.72 |
| AKT-1 | 104.50 | CDK-8/cyclin C | 97.81 |
| ALK | 99.86 | BRAFV599E | 100.92 |
| Aurora A | 48.22 | RAF-1 | 111.64 |
| Aurora B | 92.90 | MAPK APK-2 | 99.55 |
| CAMKK-2 | 83.66 | MEK-1 | 85.42 |
| VRK-2 | 118.72 | mTOR/FRAP-1 | 105.00 |
| Compound | SNB-75 (Growth%) | Compound | SNB-75 (Growth%) |
|---|---|---|---|
| 5b | 75.14 | 6d | 82.18 |
| 6a | 88.17 | 6e | 66.99 |
| 6b | 73.59 | 7a | 80.12 |
| 6c | 79.79 | 7b | 75.14 |
| Cell Cycle Phase | Positive Control (%) | Compound 6e (%) |
|---|---|---|
| G1 | 51.45 | 60.68 |
| S | 22.79 | 17.47 |
| G2 | 21.34 | 18.29 |
| Viability | Positive Control (%) | Compound 6e (%) |
|---|---|---|
| Intact cells | 98.48 | 97.83 |
| Early apoptosis | 0.08 | 0.1 |
| Late apoptosis | 0.68 | 0.81 |
| Necrosis | 0.76 | 1.26 |
| Total death | 1.52 | 2.16 |
| Compound | Docking Score (kcal/mol) | Compound | Docking Score (kcal/mol) |
|---|---|---|---|
| 5b | −11.9746 | 6d | −13.4928 |
| 6a | −11.0161 | 6e | −14.0872 |
| 6b | −12.5924 | 7a | −12.6606 |
| 6c | −16.2153 | 7b | −11.3566 |
| Binding Motif | Amino Acid * | Binding Interaction | Distance (Å) |
|---|---|---|---|
| Central motif (quinazoline) | |||
| Phenyl | Ala 160 Val 147 Leu 263 | Hydrophobic Hydrophobic Hydrophobic | - - - |
| Pyrimidine | Leu 139 Val 147 Leu 263 | Hydrophobic Hydrophobic Hydrophobic | - - - |
| F | Glu 211 Tyr 212 Ala 213 | Halogen Halogen Halogen | 3.52 2.61 2.49 |
| Terminal motif | |||
| Phenyl | Leu 139 | Hydrophobic | - |
| Br | Leu 139 Tyr 212 Pro 214 | Halogen Halogen Halogen | - - 3.27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsherbeny, M.H.; Ammar, U.M.; Abdellattif, M.H.; Abourehab, M.A.S.; Abdeen, A.; Ibrahim, S.F.; Abdelrahaman, D.; Mady, W.; Roh, E.J.; Elkamhawy, A. 2-(3-Bromophenyl)-8-fluoroquinazoline-4-carboxylic Acid as a Novel and Selective Aurora A Kinase Inhibitory Lead with Apoptosis Properties: Design, Synthesis, In Vitro and In Silico Biological Evaluation. Life 2022, 12, 876. https://doi.org/10.3390/life12060876
Elsherbeny MH, Ammar UM, Abdellattif MH, Abourehab MAS, Abdeen A, Ibrahim SF, Abdelrahaman D, Mady W, Roh EJ, Elkamhawy A. 2-(3-Bromophenyl)-8-fluoroquinazoline-4-carboxylic Acid as a Novel and Selective Aurora A Kinase Inhibitory Lead with Apoptosis Properties: Design, Synthesis, In Vitro and In Silico Biological Evaluation. Life. 2022; 12(6):876. https://doi.org/10.3390/life12060876
Chicago/Turabian StyleElsherbeny, Mohamed H., Usama M. Ammar, Magda H. Abdellattif, Mohammed A. S. Abourehab, Ahmed Abdeen, Samah F. Ibrahim, Doaa Abdelrahaman, Wessam Mady, Eun Joo Roh, and Ahmed Elkamhawy. 2022. "2-(3-Bromophenyl)-8-fluoroquinazoline-4-carboxylic Acid as a Novel and Selective Aurora A Kinase Inhibitory Lead with Apoptosis Properties: Design, Synthesis, In Vitro and In Silico Biological Evaluation" Life 12, no. 6: 876. https://doi.org/10.3390/life12060876
APA StyleElsherbeny, M. H., Ammar, U. M., Abdellattif, M. H., Abourehab, M. A. S., Abdeen, A., Ibrahim, S. F., Abdelrahaman, D., Mady, W., Roh, E. J., & Elkamhawy, A. (2022). 2-(3-Bromophenyl)-8-fluoroquinazoline-4-carboxylic Acid as a Novel and Selective Aurora A Kinase Inhibitory Lead with Apoptosis Properties: Design, Synthesis, In Vitro and In Silico Biological Evaluation. Life, 12(6), 876. https://doi.org/10.3390/life12060876