


Myostatin/AKT/FOXO Signaling Is Altered in Human Non-Ischemic Dilated Cardiomyopathy

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. RNA Isolation and cDNA Synthesis
2.3. Quantitative Real-Time Polymerase Chain Reaction
2.4. Protein Extraction and Quantification
2.5. Enzyme-Linked Immunosorbent Assays
2.6. Immunohistochemistry
2.7. Statistical Analysis
3. Results
3.1. Patient Characteristics
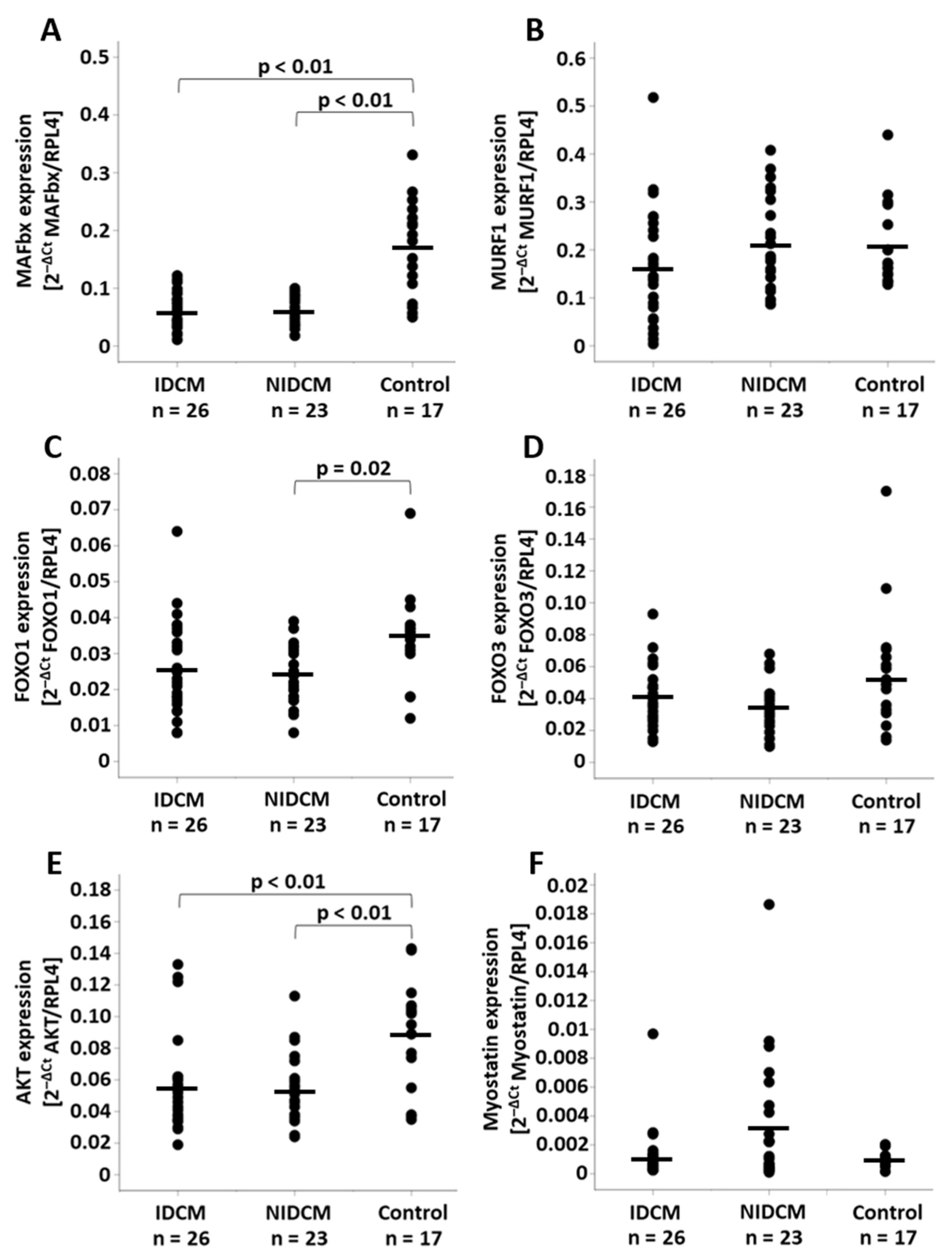
3.2. Analysis of the mRNA Expression
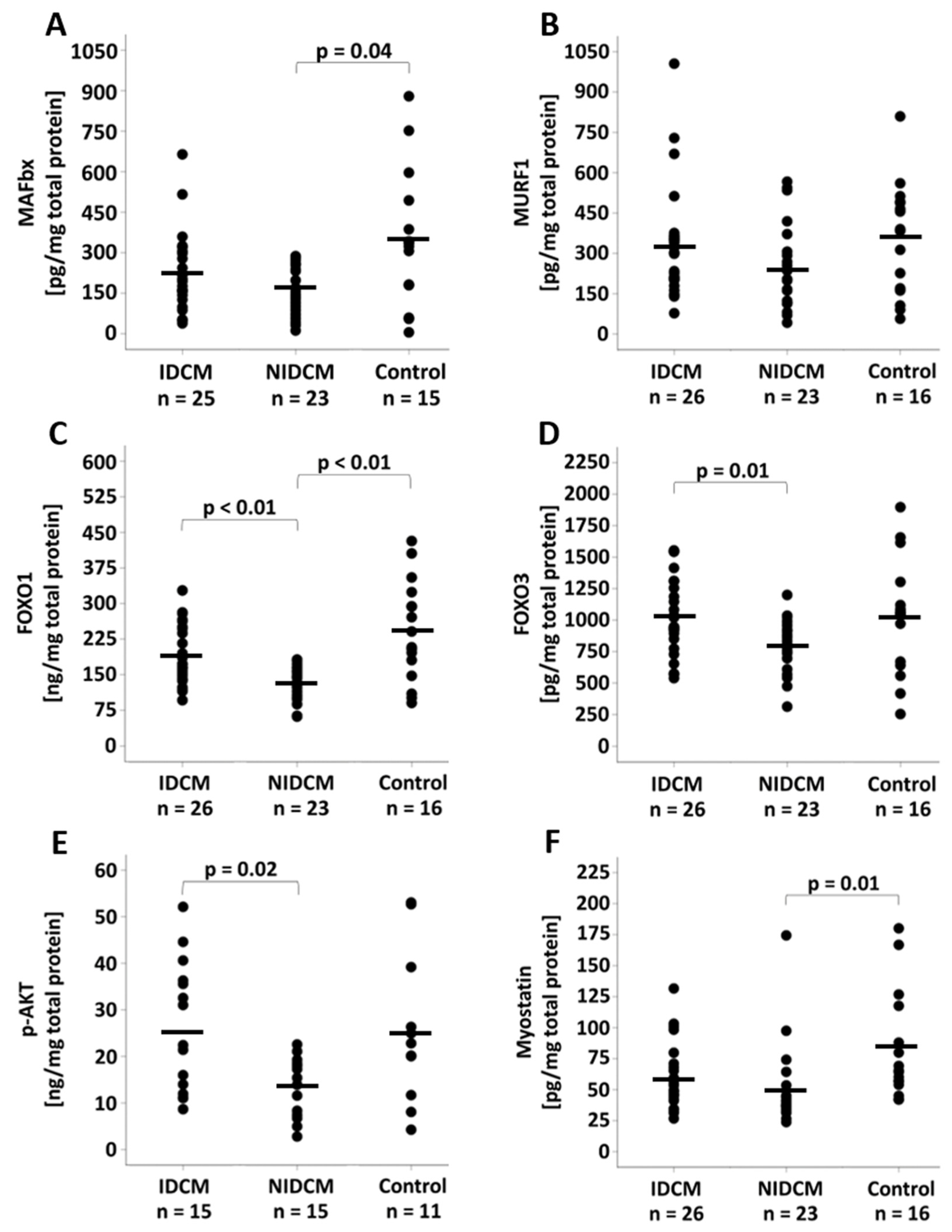
3.3. Analysis of the Protein Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drews, O.; Taegtmeyer, H. Targeting the ubiquitin-proteasome system in heart disease: The basis for new therapeutic strategies. Antioxid. Redox Signal. 2014, 21, 2322–2343. [Google Scholar] [CrossRef]
- Day, S.M. The ubiquitin proteasome system in human cardiomyopathies and heart failure. Am. J. Physiol.-Heart Circul. Physiol. 2013, 304, H1283–H1293. [Google Scholar] [CrossRef] [PubMed]
- Klaeske, K.; Dix, M.; Adams, V.; Jawad, K.; Eifert, S.; Etz, C.; Saeed, D.; Borger, M.A.; Dieterlen, M.-T. Differential Regulation of Myocardial E3 Ligases and Deubiquitinases in Ischemic Heart Failure. Life 2021, 11, 1430. [Google Scholar] [CrossRef] [PubMed]
- Skurk, C.; Izumiya, Y.; Maatz, H.; Razeghi, P.; Shiojima, I.; Sandri, M.; Sato, K.; Zeng, L.; Schiekofer, S.; Pimentel, D.; et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem. 2005, 280, 20814–20823. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol.-Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Zhang, Y.; Aguilar, O.A.; Storey, K.B. Transcriptional activation of muscle atrophy promotes cardiac muscle remodeling during mammalian hibernation. PeerJ 2016, 4, e2317. [Google Scholar] [CrossRef]
- Xin, Z.; Ma, Z.; Jiang, S.; Wang, D.; Fan, C.; Di, S.; Hu, W.; Li, T.; She, J.; Yang, Y. FOXOs in the impaired heart: New therapeutic targets for cardiac diseases. Biochim. Biophys. Acta-Mol. Basis. Dis. 2017, 1863, 486–498. [Google Scholar] [CrossRef]
- Léger, B.; Senese, R.; Al-Khodairy, A.W.; Dériaz, O.; Gobelet, C.; Giacobino, J.P.; Russell, A.P. Atrogin-1, MuRF1, and FoXO, as well as phosphorylated GSK-3beta and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord-injured patients. Muscle Nerve 2009, 40, 69–78. [Google Scholar] [CrossRef]
- Li-Li, F.; Bo-Wen, L.; Yue, X.; Zhen-Jun, T.; Meng-Xin, C. Aerobic exercise and resistance exercise alleviate skeletal muscle atrophy through IGF-1/IGF-1R-PI3K/Akt pathway in mice with myocardial infarction. Am. J. Physiol.-Cell Physiol. 2021; Epub ahead of print. [Google Scholar] [CrossRef]
- Schulze, P.C.; Fang, J.; Kassik, K.A.; Gannon, J.; Cupesi, M.; MacGillivray, C.; Lee, R.T.; Rosenthal, N. Transgenic overexpression of locally acting insulin-like growth factor-1 inhibits ubiquitin-mediated muscle atrophy in chronic left-ventricular dysfunction. Circ. Res. 2005, 97, 418–426. [Google Scholar] [CrossRef]
- Robinson, P.; Sparrow, A.J.; Patel, S.; Malinowska, M.; Reilly, S.N.; Zhang, Y.H.; Casadei, B.; Watkins, H.; Redwood, C. Dilated cardiomyopathy mutations in thin-filament regulatory proteins reduce contractility, suppress systolic Ca2+, and activate NFAT and Akt signaling. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H306–H319. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Naito, A.T.; Higo, T.; Nakagawa, A.; Shibamoto, M.; Sakai, T.; Hashimoto, A.; Kuramoto, Y.; Sumida, T.; Nomura, S.; et al. Wnt/β-Catenin Signaling Contributes to Skeletal Myopathy in Heart Failure via Direct Interaction With Forkhead Box O. Circ. Heart Fail. 2015, 8, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.R.; Cook, S.A.; Foo, S.; McKoy, G.; Ashida, N.; Novikov, M.; Scherrer-Crosbie, M.; Li, L.; Matsui, T.; Brooks, G.; et al. Myostatin regulates cardiomyocyte growth through modulation of Akt signaling. Circ. Res. 2006, 99, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Battiprolu, P.K.; Hojayev, B.; Jiang, N.; Wang, Z.V.; Luo, X.; Iglewski, M.; Shelton, J.M.; Gerard, R.D.; Rothermel, B.A.; Gillette, T.G.; et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Investig. 2012, 122, 1109–1118. [Google Scholar] [CrossRef]
- Razeghi, P.; Baskin, K.K.; Sharma, S.; Young, M.E.; Stepkowski, S.; Essop, M.F.; Taegtmeyer, H. Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem. Biophys. Res. Commun. 2006, 342, 361–364. [Google Scholar] [CrossRef]
- Baskin, K.K.; Rodriguez, M.R.; Kansara, S.; Chen, W.; Carranza, S.; Frazier, O.H.; Glass, D.J.; Taegtmeyer, H. MAFbx/Atrogin-1 is required for atrophic remodeling of the unloaded heart. J. Mol. Cell. Cardiol. 2014, 72, 168–176. [Google Scholar] [CrossRef]
- Yu, W.; Chen, C.; Cheng, J. The role and molecular mechanism of FoxO1 in mediating cardiac hypertrophy. ESC Heart Fail. 2020, 7, 3497–3504. [Google Scholar] [CrossRef]
- Qi, Y.; Zhu, Q.; Zhang, K.; Thomas, C.; Wu, Y.; Kumar, R.; Baker, K.M.; Xu, Z.; Chen, S.; Guo, S. Activation of Foxo1 by insulin resistance promotes cardiac dysfunction and β-myosin heavy chain gene expression. Circ. Heart Fail. 2015, 8, 198–208. [Google Scholar] [CrossRef]
- Razeghi, P.; Taegtmeyer, H. Hypertrophy and atrophy of the heart: The other side of remodeling. Ann. N. Y. Acad. Sci. 2006, 1080, 110–119. [Google Scholar] [CrossRef]
- Kostin, S.; Pool, L.; Elsässer, A.; Hein, S.; Drexler, H.C.; Arnon, E.; Hayakawa, Y.; Zimmermann, R.; Bauer, E.; Klövekorn, W.P.; et al. Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 2003, 92, 715–724. [Google Scholar] [CrossRef]
- Weekes, J.; Morrison, K.; Mullen, A.; Wait, R.; Barton, P.; Dunn, M.J. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 2003, 3, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Day, S.M.; Divald, A.; Wang, P.; Davis, F.; Bartolone, S.; Jones, R.; Powell, S.R. Impaired assembly and post-translational regulation of 26S proteasome in human end-stage heart failure. Circ. Heart Fail. 2013, 6, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Wohlschlaeger, J.; Sixt, S.U.; Stoeppler, T.; Schmitz, K.J.; Levkau, B.; Tsagakis, K.; Vahlhaus, C.; Schmid, C.; Peters, J.; Schmid, K.W.; et al. Ventricular unloading is associated with increased 20 s proteasome protein expression in the myocardium. J. Heart Lung Transplant. 2010, 29, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Spänig, S.; Kellermann, K.; Dieterlen, M.T.; Noack, T.; Lehmann, S.; Borger, M.A.; Garbade, J.; Barac, Y.D.; Emrich, F. The Ubiquitin Proteasome System in Ischemic and Dilated Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 6354. [Google Scholar] [CrossRef]
- Galasso, G.; De Rosa, R.; Piscione, F.; Iaccarino, G.; Vosa, C.; Sorriento, D.; Piccolo, R.; Rapacciuolo, A.; Walsh, K.; Chiariello, M. Myocardial expression of FOXO3a-Atrogin-1 pathway in human heart failure. Eur. J. Heart Fail. 2010, 12, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Conraads, V.M.; Vrints, C.J.; Rodrigus, I.E.; Hoymans, V.Y.; Van Craenenbroeck, E.M.; Bosmans, J.; Claeys, M.J.; Van Herck, P.; Linke, A.; Schuler, G.; et al. Depressed expression of MuRF1 and MAFbx in areas remote of recent myocardial infarction: A mechanism contributing to myocardial remodeling? Basic Res. Cardiol. 2010, 105, 219–226. [Google Scholar] [CrossRef]
- Perry, B.D.; Caldow, M.K.; Brennan-Speranza, T.C.; Sbaraglia, M.; Jerums, G.; Garnham, A.; Wong, C.; Levinger, P.; Asrar Ul Haq, M.; Hare, D.L.; et al. Muscle atrophy in patients with Type 2 Diabetes Mellitus: Roles of inflammatory pathways, physical activity and exercise. Exerc. Immunol. Rev. 2016, 22, 94–109. [Google Scholar]
- Castillero, E.; Ali, Z.A.; Akashi, H.; Giangreco, N.; Wang, C.; Stöhr, E.J.; Ji, R.; Zhang, X.; Kheysin, N.; Park, J.S.; et al. Structural and functional cardiac profile after prolonged duration of mechanical unloading: Potential implications for myocardial recovery. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1463–H1476. [Google Scholar] [CrossRef]
- George, I.; Bish, L.T.; Kamalakkannan, G.; Petrilli, C.M.; Oz, M.C.; Naka, Y.; Sweeney, H.L.; Maybaum, S. Myostatin activation in patients with advanced heart failure and after mechanical unloading. Eur. J. Heart Fail. 2010, 12, 444–453. [Google Scholar] [CrossRef]
- Topkara, V.K.; Garan, A.R.; Fine, B.; Godier-Furnémont, A.F.; Breskin, A.; Cagliostro, B.; Yuzefpolskaya, M.; Takeda, K.; Takayama, H.; Mancini, D.M.; et al. Myocardial Recovery in Patients Receiving Contemporary Left Ventricular Assist Devices: Results From the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS). Circ. Heart Fail. 2016, 9, e003157. [Google Scholar] [CrossRef] [Green Version]
- Antonides, C.F.J.; Schoenrath, F.; de By, T.M.M.H.; Muslem, R.; Veen, K.; Yalcin, Y.C.; Netuka, I.; Gummert, J.; Potapov, E.V.; Meyns, B.; et al. EUROMACS investigators. Outcomes of patients after successful left ventricular assist device explantation: A EUROMACS study. ESC Heart Fail. 2020, 7, 1085–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| IDCM (n = 26) | NIDCM (n = 23) | Control (n = 17) | p Value | |
|---|---|---|---|---|
| Male sex | 17 (65.4%) | 12 (52.2%) | 8 (47.1%) | 0.45 |
| Age at surgical intervention [years] | 65.5 ± 6.4 | 66.7 ± 8.3 | 69.2 ± 7.9 | 0.28 |
| BMI [kg/m2] | 28.2 ± 5.4 | 27.5 ± 6.0 | 30.2 ± 4.8 | 0.29 |
| LVEF [%] | 22.0 ± 6.7 | 21.6 ± 5.3 | 63.1 ± 9.7 | <0.01 |
| NT-proBNP [ng/L] | 9042 ± 8996 | 10,488 ± 16,700 | - | 0.36 |
| NYHA classification | <0.01 | |||
| Class I | 1 (3.8%) | 0 (0%) | 4 (23.5%) | |
| Class II | 3 (11.5%) | 1 (4.3%) | 8 (47.1%) | |
| Class III | 7 (26.9%) | 17 (73.9%) | 6 (29.4%) | |
| Class IV | 15 (57.7%) | 5 (21.7%) | 0 (0%) | |
| Nicotine abuse | 0.07 | |||
| Smoker | 8 (30.8%) | 2 (8.7%) | 0 (0.0%) | |
| Non-smoker | 12 (46.2%) | 13 (56.5%) | 13 (76.5%) | |
| Ex-smoker | 5 (19.2%) | 8 (34.8%) | 4 (23.5%) | |
| Unknown | 1 (3.8%) | 0 (0%) | 0 (0%) | |
| Arterial hypertension | 24 (92.3%) | 16 (69.6%) | 13 (76.5%) | 0.12 |
| Coronary heart disease | 26 (100%) | 3 (13.0%) | 5 (29.4%) | <0.01 |
| Type 2 diabetes | 15 (57.7%) | 7 (30.4%) | 3 (17.6%) | 0.02 |
| Myocardial infarction | 24 (92.3%) | 0 (0%) | 0 (0%) | <0.01 |
| Renal function eGFR [mL/min/1.73 m2] Creatinine [µmol/L] | 43.9 ± 19.7 159.4 ± 80.1 | 45.1 ± 20.8 158.2 ± 112.5 | 78.1 ± 17.9 79.3 ± 27.2 | <0.01 <0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hildebrandt, L.; Dieterlen, M.-T.; Klaeske, K.; Haunschild, J.; Saeed, D.; Eifert, S.; Borger, M.A.; Jawad, K. Myostatin/AKT/FOXO Signaling Is Altered in Human Non-Ischemic Dilated Cardiomyopathy. Life 2022, 12, 1418. https://doi.org/10.3390/life12091418
Hildebrandt L, Dieterlen M-T, Klaeske K, Haunschild J, Saeed D, Eifert S, Borger MA, Jawad K. Myostatin/AKT/FOXO Signaling Is Altered in Human Non-Ischemic Dilated Cardiomyopathy. Life. 2022; 12(9):1418. https://doi.org/10.3390/life12091418
Chicago/Turabian StyleHildebrandt, Lea, Maja-Theresa Dieterlen, Kristin Klaeske, Josephina Haunschild, Diyar Saeed, Sandra Eifert, Michael A. Borger, and Khalil Jawad. 2022. "Myostatin/AKT/FOXO Signaling Is Altered in Human Non-Ischemic Dilated Cardiomyopathy" Life 12, no. 9: 1418. https://doi.org/10.3390/life12091418
APA StyleHildebrandt, L., Dieterlen, M. -T., Klaeske, K., Haunschild, J., Saeed, D., Eifert, S., Borger, M. A., & Jawad, K. (2022). Myostatin/AKT/FOXO Signaling Is Altered in Human Non-Ischemic Dilated Cardiomyopathy. Life, 12(9), 1418. https://doi.org/10.3390/life12091418