
What Links Chronic Kidney Disease and Ischemic Cardiomyopathy? A Comprehensive Bioinformatic Analysis Utilizing Bulk and Single-Cell RNA Sequencing Data with Machine Learning

Abstract
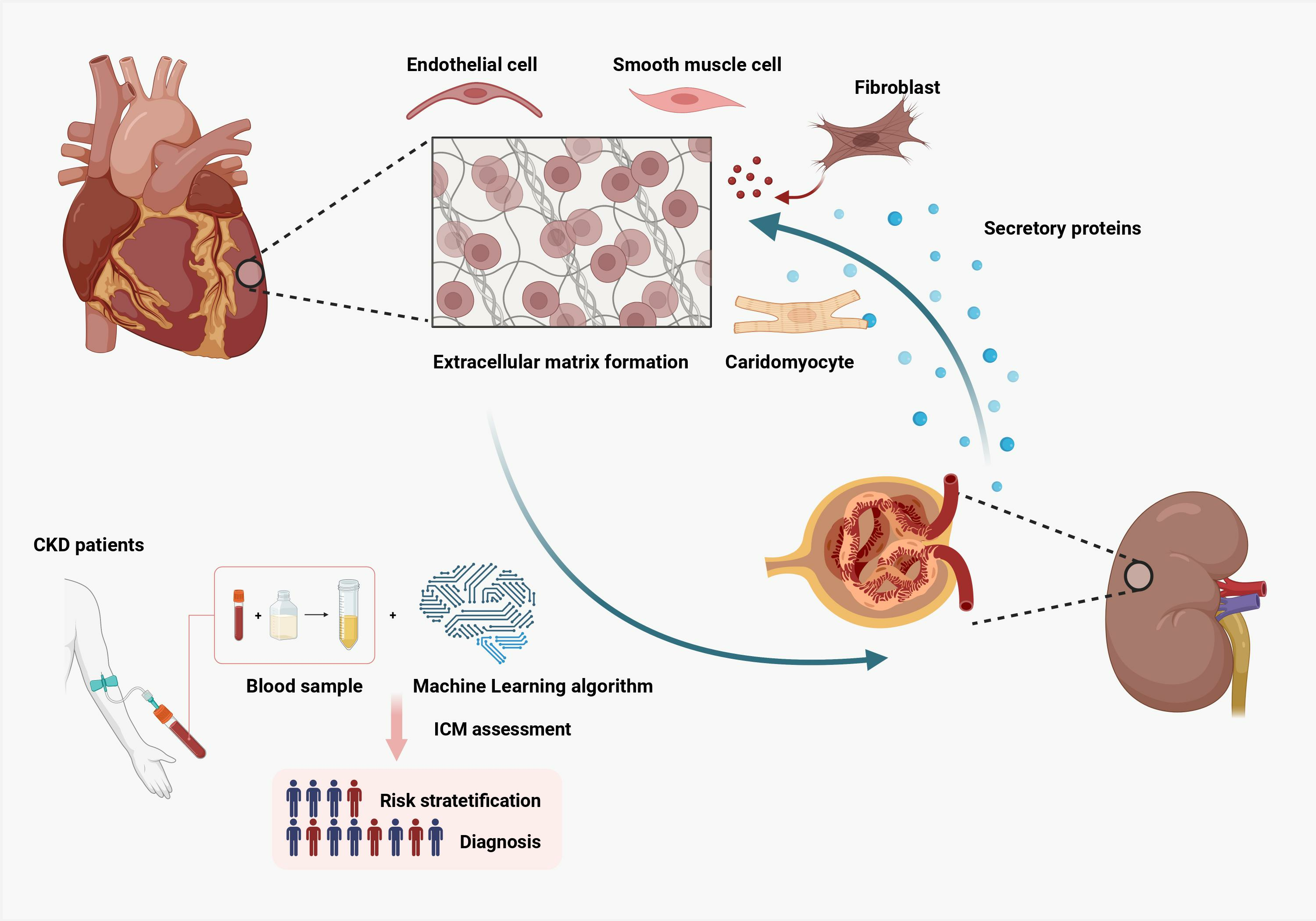
:
1. Introduction
2. Methods
2.1. Data Collection
2.2. Differentially Expressed Genes (DEGs) Analysis
2.3. Functional Enrichment Analysis
2.4. Protein–Protein Interaction (PPI) Network Construction
2.5. Secretory Proteins Access
2.6. Feature Selection Based on Machine Learning
2.7. scRNA-Seq Data Analysis
2.8. Classifier Construction and Assessment Based on Machine Learning Algorithm
2.9. Statistical Analysis
3. Results
3.1. Identification of DEGs in ICM and CKD and Functional Enrichment Analysis
3.2. Identification of Candidate Genes for CKD-Related ICM
3.3. scRNA-Sequencing Analysis of ICM
3.4. Construction and Validation of a Diagnostic Model for CKD-Related ICM Using Machine Learning Algorithms
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease-A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef]
- Schuett, K.; Marx, N.; Lehrke, M. The cardio-kidney patient: Epidemiology, clinical characteristics and therapy. Circ. Res. 2023, 132, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Sarnak, M.J.; Amann, K.; Bangalore, S.; Cavalcante, J.L.; Charytan, D.M.; Craig, J.C.; Gill, J.S.; Hlatky, M.A.; Jardine, A.G.; Landmesser, U. Chronic kidney disease and coronary artery disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2019, 74, 1823–1838. [Google Scholar] [CrossRef] [PubMed]
- Hage, F.G.; Venkataraman, R.; Zoghbi, G.J.; Perry, G.J.; DeMattos, A.M.; Iskandrian, A.E. The scope of coronary heart disease in patients with chronic kidney disease. J. Am. Coll. Cardiol. 2009, 53, 2129–2140. [Google Scholar] [CrossRef]
- Chinnappa, S.; White, E.; Lewis, N.; Baldo, O.; Tu, Y.-K.; Glorieux, G.; Vanholder, R.; El Nahas, M.; Mooney, A. Early and asymptomatic cardiac dysfunction in chronic kidney disease. Nephrol. Dial. Transplant. 2018, 33, 450–458. [Google Scholar] [CrossRef]
- Cai, Q.; K Mukku, V.; Ahmad, M. Coronary artery disease in patients with chronic kidney disease: A clinical update. Curr. Cardiol. Rev. 2013, 9, 331–339. [Google Scholar] [CrossRef]
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular disease in chronic kidney disease: Pathophysiological insights and therapeutic options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef]
- Jia, T.; Olauson, H.; Lindberg, K.; Amin, R.; Edvardsson, K.; Lindholm, B.; Andersson, G.; Wernerson, A.; Sabbagh, Y.; Schiavi, S. A novel model of adenine-induced tubulointerstitial nephropathy in mice. BMC Nephrol. 2013, 14, 116. [Google Scholar] [CrossRef]
- Fularski, P.; Krzemińska, J.; Lewandowska, N.; Młynarska, E.; Saar, M.; Wronka, M.; Rysz, J.; Franczyk, B. Statins in Chronic Kidney Disease—Effects on Atherosclerosis and Cellular Senescence. Cells 2023, 12, 1679. [Google Scholar] [CrossRef]
- Yan, C.; Xu, Z.; Huang, W. Cellular senescence affects cardiac regeneration and repair in ischemic heart disease. Aging Dis. 2021, 12, 552. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.R.; Robbins, M.J.; Kim, M.C.; Fuster, V. Management of cardiovascular disease in patients with kidney disease. Nat. Rev. Cardiol. 2013, 10, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Dilsizian, V.; Gewirtz, H.; Marwick, T.H.; Kwong, R.Y.; Raggi, P.; Al-Mallah, M.H.; Herzog, C.A. Cardiac imaging for coronary heart disease risk stratification in chronic kidney disease. Cardiovasc. Imaging 2021, 14, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis; Taylor & Francis: Abingdon, UK, 2019. [Google Scholar]
- Gu, Z. Complex heatmap visualization. Imeta 2022, 1, e43. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2016, 060012. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1. [Google Scholar] [CrossRef]
- Kursa, M.B.; Rudnicki, W.R. Feature selection with the Boruta package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef]
- RColorBrewer, S.; Liaw, M.A. Package ‘Randomforest’; University of California, Berkeley: Berkeley, CA, USA, 2018. [Google Scholar]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-r.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef]
- Franzén, O.; Gan, L.-M.; Björkegren, J.L. PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.M.; Villar, K.N.B.; Small, E.M. Fibroblast contributions to ischemic cardiac remodeling. Cell. Signal. 2021, 77, 109824. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Nohara, Y.; Matsumoto, K.; Soejima, H.; Nakashima, N. Explanation of machine learning models using improved shapley additive explanation. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA, 7–10 September 2019; p. 546. [Google Scholar]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef]
- Maruyama, K.; Imanaka-Yoshida, K. The pathogenesis of cardiac fibrosis: A review of recent progress. Int. J. Mol. Sci. 2022, 23, 2617. [Google Scholar] [CrossRef]
- Voss, S.; Krüger, S.; Scherschel, K.; Warnke, S.; Schwarzl, M.; Schrage, B.; Girdauskas, E.; Meyer, C.; Blankenberg, S.; Westermann, D. Macrophage migration inhibitory factor (MIF) expression increases during myocardial infarction and supports pro-inflammatory signaling in cardiac fibroblasts. Biomolecules 2019, 9, 38. [Google Scholar] [CrossRef]
- Chintalgattu, V.; Nair, D.M.; Katwa, L.C. Cardiac myofibroblasts: A novel source of vascular endothelial growth factor (VEGF) and its receptors Flt-1 and KDR. J. Mol. Cell. Cardiol. 2003, 35, 277–286. [Google Scholar] [CrossRef]
- Johnson, T.; Zhao, L.; Manuel, G.; Taylor, H.; Liu, D. Approaches to therapeutic angiogenesis for ischemic heart disease. J. Mol. Med. 2019, 97, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.R.; Balaji, S.; Narmoneva, D.A. Complex temporal regulation of capillary morphogenesis by fibroblasts. Am. J. Physiol.-Cell Physiol. 2010, 299, C444–C453. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, M.; Tang, X.; Huang, J.; Li, J.; Hong, X.; Fu, H.; Liu, Y. Proteomic landscape of the extracellular matrix in the fibrotic kidney. Kidney Int. 2023, 103, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Decharatchakul, N.; Settasatian, C.; Settasatian, N.; Komanasin, N.; Kukongviriyapan, U.; Intharapetch, P.; Senthong, V.; Sawanyawisuth, K. Association of combined genetic variations in SOD3, GPX3, PON1, and GSTT1 with hypertension and severity of coronary artery disease. Heart Vessel. 2020, 35, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Pang, P.; Abbott, M.; Abdi, M.; Fucci, Q.-A.; Chauhan, N.; Mistri, M.; Proctor, B.; Chin, M.; Wang, B.; Yin, W. Pre-clinical model of severe glutathione peroxidase-3 deficiency and chronic kidney disease results in coronary artery thrombosis and depressed left ventricular function. Nephrol. Dial. Transplant. 2018, 33, 923–934. [Google Scholar] [CrossRef]
- Li, G.; Qin, Y.; Cheng, Z.; Cheng, X.; Wang, R.; Luo, X.; Zhao, Y.; Zhang, D.; Li, G. Gpx3 and Egr1 are involved in regulating the differentiation fate of cardiac fibroblasts under pressure overload. Oxidative Med. Cell. Longev. 2022, 2022, 3235250. [Google Scholar] [CrossRef]
- Galasso, G.; Schiekofer, S.; Sato, K.; Shibata, R.; Handy, D.E.; Ouchi, N.; Leopold, J.A.; Loscalzo, J.; Walsh, K. Impaired angiogenesis in glutathione peroxidase-1–deficient mice is associated with endothelial progenitor cell dysfunction. Circ. Res. 2006, 98, 254–261. [Google Scholar] [CrossRef]
- Calabro, N.E.; Kristofik, N.J.; Kyriakides, T.R. Thrombospondin-2 and extracellular matrix assembly. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 2396–2402. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Liu, I.-F.; Kuo, H.-F.; Li, C.-Y.; Lian, W.-S.; Chang, C.-Y.; Chen, Y.-H.; Liu, W.-L.; Lu, C.-Y.; Liu, Y.-R. miR-29a-3p/THBS2 axis regulates PAH-induced cardiac fibrosis. Int. J. Mol. Sci. 2021, 22, 10574. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, M.; Cao, D.; Zheng, A.; Zhang, Y.; Su, Y.; Wang, J.; Xu, Y.; Zhou, M.; Tang, Y. Inhibition of Fap Promotes Cardiac Repair by Stabilizing BNP. Circ. Res. 2023, 132, 586–600. [Google Scholar] [CrossRef]
- Christman, K.L.; Fang, Q.; Yee, M.S.; Johnson, K.R.; Sievers, R.E.; Lee, R.J. Enhanced neovasculature formation in ischemic myocardium following delivery of pleiotrophin plasmid in a biopolymer. Biomaterials 2005, 26, 1139–1144. [Google Scholar] [CrossRef]
- Yokoi, H.; Kasahara, M.; Mori, K.; Ogawa, Y.; Kuwabara, T.; Imamaki, H.; Kawanishi, T.; Koga, K.; Ishii, A.; Kato, Y. Pleiotrophin triggers inflammation and increased peritoneal permeability leading to peritoneal fibrosis. Kidney Int. 2012, 81, 160–169. [Google Scholar] [CrossRef] [PubMed]
- De Lemos, J.A.; Hillis, L.D. Diagnosis and management of coronary artery disease in patients with end-stage renal disease on hemodialysis. J. Am. Soc. Nephrol. 1996, 7, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Tsukamoto, K.; Hasebe, N.; Tamaki, N.; Kikuchi, K.; Ono, T. Prediction of cardiac death in hemodialysis patients by myocardial fatty acid imaging. J. Am. Coll. Cardiol. 2008, 51, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, D.J.; Zhang, X.H.; Leano, R.; Armstrong, K.A.; Isbel, N.M.; Marwick, T.H. Prognostic role of subclinical left ventricular abnormalities and impact of transplantation in chronic kidney disease. Am. Heart J. 2007, 153, 656–664. [Google Scholar] [CrossRef]
- Edwards, N.C.; Moody, W.E.; Chue, C.D.; Ferro, C.J.; Townend, J.N.; Steeds, R.P. Defining the natural history of uremic cardiomyopathy in chronic kidney disease: The role of cardiovascular magnetic resonance. JACC Cardiovasc. Imaging 2014, 7, 703–714. [Google Scholar] [CrossRef]
- Peng, W.K.; Chen, L.; Boehm, B.O.; Han, J.; Loh, T.P. Molecular phenotyping of oxidative stress in diabetes mellitus with point-of-care NMR system. npj Aging Mech. Dis. 2020, 6, 11. [Google Scholar] [CrossRef]
- Peng, W.K.; Ng, T.-T.; Loh, T.P. Machine learning assistive rapid, label-free molecular phenotyping of blood with two-dimensional NMR correlational spectroscopy. Commun. Biol. 2020, 3, 535. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession | Platform | Origin | Sample | Species | |
|---|---|---|---|---|---|
| Control | Disease | ||||
| GSE5406 | GPL96 | Heart | 16 | 108 | Homo Sapiens |
| GSE37171 | GPL570 | PBMC | 40 | 75 | Homo Sapiens |
| GSE57345 | GPL11532 | Heart | 136 | 95 | Homo Sapiens |
| GSE145154 | GPL20795 | Heart | 5 | 14 | Homo Sapiens |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Chen, Y.; Huang, W. What Links Chronic Kidney Disease and Ischemic Cardiomyopathy? A Comprehensive Bioinformatic Analysis Utilizing Bulk and Single-Cell RNA Sequencing Data with Machine Learning. Life 2023, 13, 2215. https://doi.org/10.3390/life13112215
Yang L, Chen Y, Huang W. What Links Chronic Kidney Disease and Ischemic Cardiomyopathy? A Comprehensive Bioinformatic Analysis Utilizing Bulk and Single-Cell RNA Sequencing Data with Machine Learning. Life. 2023; 13(11):2215. https://doi.org/10.3390/life13112215
Chicago/Turabian StyleYang, Lingzhi, Yunwei Chen, and Wei Huang. 2023. "What Links Chronic Kidney Disease and Ischemic Cardiomyopathy? A Comprehensive Bioinformatic Analysis Utilizing Bulk and Single-Cell RNA Sequencing Data with Machine Learning" Life 13, no. 11: 2215. https://doi.org/10.3390/life13112215
APA StyleYang, L., Chen, Y., & Huang, W. (2023). What Links Chronic Kidney Disease and Ischemic Cardiomyopathy? A Comprehensive Bioinformatic Analysis Utilizing Bulk and Single-Cell RNA Sequencing Data with Machine Learning. Life, 13(11), 2215. https://doi.org/10.3390/life13112215