Genome-Based Classification and Therapy of Prostate Cancer



Abstract
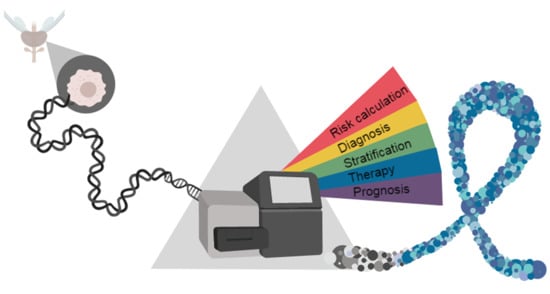
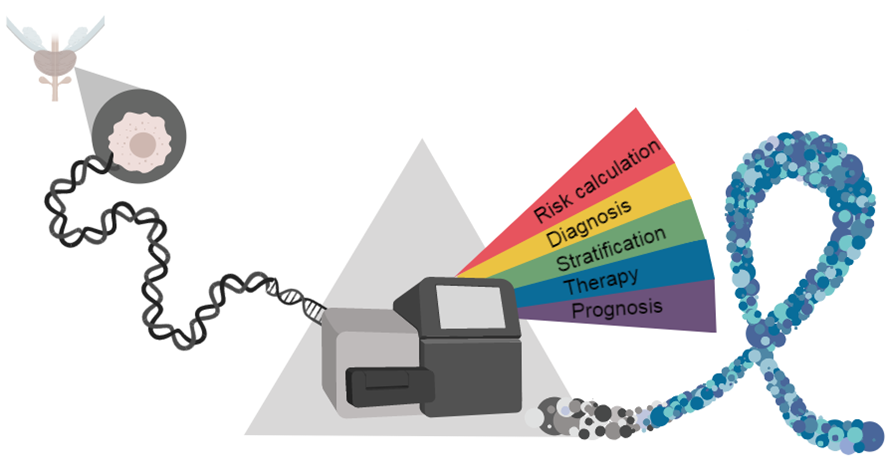
:
1. Introduction
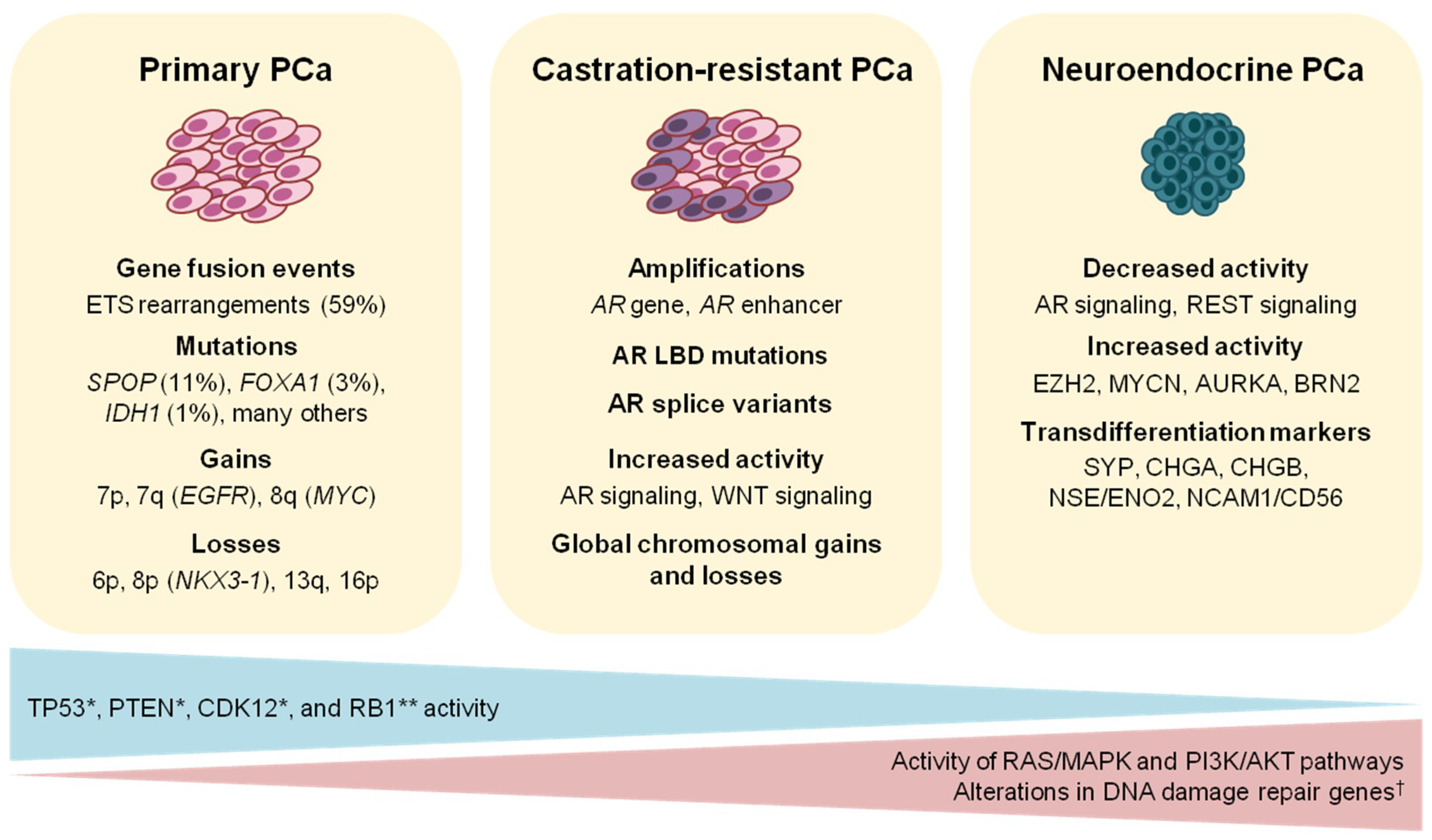
2. The Genomic Landscape of Primary Prostate Cancer
2.1. ETS Fusion-Positive Prostate Cancer
2.2. ETS Fusion-Negative Prostate Cancer
2.3. Cellular Processes Deregulated in Prostate Cancer
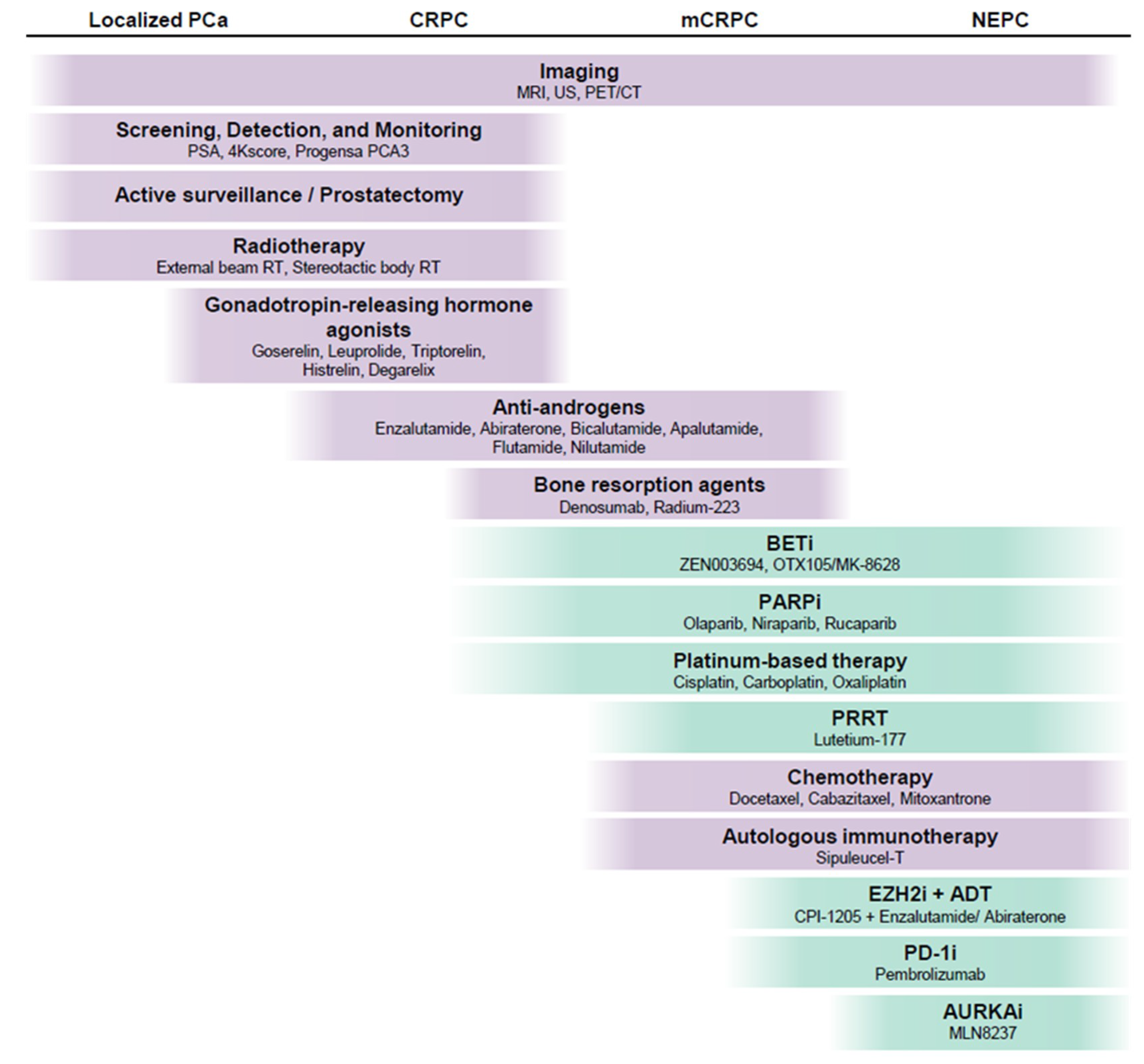
3. Current Molecular Biomarkers for Prostate Cancer Diagnosis and Risk Stratification
4. Prognostic Molecular Marker Signature in Prostate Cancer
5. Treatment of Localized Prostate Cancer
6. Molecular Alterations and Current Therapy of Advanced Prostate Cancer
Androgen Deprivation Therapy and Combined Androgen Blockade in Metastatic Prostate Cancer
7. Novel Approaches to the Classification and Treatment of Advanced Prostate Cancer
7.1. DNA Repair
7.2. Immune Checkpoint Inhibition
7.3. Epigenetic Alterations
7.4. Molecular Markers of Neuroendocrine Prostate Cancer
8. Summary
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- SEER Cancer Statistics Review, 1975–2014. Available online: https://seer.cancer.gov/csr/1975_2014/ (accessed on 30 April 2018).
- Lee, J.; Demissie, K.; Lu, S.E.; Rhoads, G.G. Cancer incidence among Korean—American immigrants in the United States and native Koreans in South Korea. Cancer Control 2007, 14, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Zeigler-Johnson, C.M.; Rennert, H.; Mittal, R.D.; Jalloh, M.; Sachdeva, R.; Malkowicz, S.B.; Mandhani, A.; Mittal, B.; Gueye, S.M.; Rebbeck, T.R. Evaluation of prostate cancer characteristics in four populations worldwide. Can. J. Urol. 2008, 15, 4056–4064. [Google Scholar] [PubMed]
- Thompson, I.M.; Pauler, D.K.; Goodman, P.J.; Tangen, C.M.; Lucia, M.S.; Parnes, H.L.; Minasian, L.M.; Ford, L.G.; Lippman, S.M.; Crawford, E.D.; et al. Prevalence of prostate cancer among men with a prostate-specific antigen level ≤4.0 ng per milliliter. N. Engl. J. Med. 2004, 350, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Eeles, R.A.; Kote-Jarai, Z.; Al Olama, A.A.; Giles, G.G.; Guy, M.; Severi, G.; Muir, K.; Hopper, J.L.; Henderson, B.E.; Haiman, C.A.; et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat. Genet. 2009, 41, 1116–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, F.R.; Berndt, S.I.; Siddiq, A.; Jacobs, K.B.; Wang, Z.; Lindstrom, S.; Stevens, V.L.; Chen, C.; Mondul, A.M.; Travis, R.C.; et al. Genome-wide association study identifies new prostate cancer susceptibility loci. Hum. Mol. Genet. 2011, 20, 3867–3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haiman, C.A.; Chen, G.K.; Blot, W.J.; Strom, S.S.; Berndt, S.I.; Kittles, R.A.; Rybicki, B.A.; Isaacs, W.B.; Ingles, S.A.; Stanford, J.L.; et al. Genome-wide association study of prostate cancer in men of African ancestry identifies a susceptibility locus at 17q21. Nat. Genet. 2011, 43, 570–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kote-Jarai, Z.; Olama, A.A.; Giles, G.G.; Severi, G.; Schleutker, J.; Weischer, M.; Campa, D.; Riboli, E.; Key, T.; Gronberg, H.; et al. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat. Genet. 2011, 43, 785–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Fakhrejahani, F.; Madan, R.A.; Dahut, W.L. Management Options for Biochemically Recurrent Prostate Cancer. Curr. Treat. Options Oncol. 2017, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Mahon, K.L.; Henshall, S.M.; Sutherland, R.L.; Horvath, L.G. Pathways of chemotherapy resistance in castration-resistant prostate cancer. Endocr. Relat. Cancer 2011, 18, R103–R123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galazi, M.; Rodriguez-Vida, A.; Ng, T.; Mason, M.; Chowdhury, S. Precision medicine for prostate cancer. Expert Rev. Anticancer Ther. 2014, 14, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Rubin, M.A.; Demichelis, F. The Genomics of Prostate Cancer: Emerging understanding with technologic advances. Mod. Pathol. 2018, 31, S1–S11. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hieronymus, H.; Schultz, N.; Gopalan, A.; Carver, B.S.; Chang, M.T.; Xiao, Y.; Heguy, A.; Huberman, K.; Bernstein, M.; Assel, M.; et al. Copy number alteration burden predicts prostate cancer relapse. Proc. Natl. Acad. Sci. USA 2014, 111, 11139–11144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Simon, R.; Feuerbach, L.; Schlangen, K.; Weichenhan, D.; Minner, S.; Wuttig, D.; Warnatz, H.J.; Stehr, H.; Rausch, T.; et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 2013, 23, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.G.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Tran, J.; Gopalan, A.; Chen, Z.; Shaikh, S.; Carracedo, A.; Alimonti, A.; Nardella, C.; Varmeh, S.; Scardino, P.T.; et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat. Genet. 2009, 41, 619–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 2009, 41, 524–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickman, D.S.; Chen, Y.B.; Banerjee, S.; Pan, Y.; Yu, J.; Vuong, T.; Perner, S.; Lafargue, C.J.; Mertz, K.D.; Setlur, S.R.; et al. ERG cooperates with androgen receptor in regulating trefoil factor 3 in prostate cancer disease progression. Neoplasia 2010, 12, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Ratz, L.; Laible, M.; Kacprzyk, L.A.; Wittig-Blaich, S.M.; Tolstov, Y.; Duensing, S.; Altevogt, P.; Klauck, S.M.; Sultmann, H. TMPRSS2:ERG gene fusion variants induce TGF-beta signaling and epithelial to mesenchymal transition in human prostate cancer cells. Oncotarget 2017, 8, 25115–25130. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.J.; Fraser, M.; van der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Soong, T.D.; Moss, B.; Mosquera, J.M.; Dlabal, J.; Terry, S.; MacDonald, T.Y.; Tripodi, J.; Bunting, K.; Najfeld, V.; et al. Oncogene-mediated alterations in chromatin conformation. Proc. Natl. Acad. Sci. USA 2012, 109, 9083–9088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demichelis, F.; Fall, K.; Perner, S.; Andren, O.; Schmidt, F.; Setlur, S.R.; Hoshida, Y.; Mosquera, J.M.; Pawitan, Y.; Lee, C.; et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 2007, 26, 4596–4599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attard, G.; Clark, J.; Ambroisine, L.; Fisher, G.; Kovacs, G.; Flohr, P.; Berney, D.; Foster, C.S.; Fletcher, A.; Gerald, W.L.; et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 2008, 27, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Aubin, S.M.; Siddiqui, J.; Lonigro, R.J.; Sefton-Miller, L.; Miick, S.; Williamsen, S.; Hodge, P.; Meinke, J.; Blase, A.; et al. Urine TMPRSS2:ERG fusion transcript stratifies prostate cancer risk in men with elevated serum PSA. Sci. Transl. Med. 2011, 3, 94ra72. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, A.; Leversha, M.A.; Satagopan, J.M.; Zhou, Q.; Al-Ahmadie, H.A.; Fine, S.W.; Eastham, J.A.; Scardino, P.T.; Scher, H.I.; Tickoo, S.K.; et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res. 2009, 69, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Fine, S.W.; Gopalan, A.; Leversha, M.A.; Al-Ahmadie, H.A.; Tickoo, S.K.; Zhou, Q.; Satagopan, J.M.; Scardino, P.T.; Gerald, W.L.; Reuter, V.E. TMPRSS2-ERG gene fusion is associated with low Gleason scores and not with high-grade morphological features. Mod. Pathol. 2010, 23, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- McKiernan, J.; Donovan, M.J.; O’Neill, V.; Bentink, S.; Noerholm, M.; Belzer, S.; Skog, J.; Kattan, M.W.; Partin, A.; Andriole, G.; et al. A Novel Urine Exosome Gene Expression Assay to Predict High-grade Prostate Cancer at Initial Biopsy. JAMA Oncol. 2016, 2, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.; Torkler, P.; Skog, J.; Noerholm, M.; Carroll, P. Performance of a validated urine exosome gene expression assay to predict high-grade prostate cancer utilizing the International Society of Urological Pathology (ISUP) 2014 grading system. J. Clin. Oncol. 2017, 35, 49. [Google Scholar] [CrossRef]
- Cornu, J.N.; Cancel-Tassin, G.; Egrot, C.; Gaffory, C.; Haab, F.; Cussenot, O. Urine TMPRSS2:ERG fusion transcript integrated with PCA3 score, genotyping, and biological features are correlated to the results of prostatic biopsies in men at risk of prostate cancer. Prostate 2013, 73, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Leyten, G.H.; Hessels, D.; Jannink, S.A.; Smit, F.P.; de Jong, H.; Cornel, E.B.; de Reijke, T.M.; Vergunst, H.; Kil, P.; Knipscheer, B.C.; et al. Prospective multicentre evaluation of PCA3 and TMPRSS2-ERG gene fusions as diagnostic and prognostic urinary biomarkers for prostate cancer. Eur. Urol. 2014, 65, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Kulda, V.; Topolcan, O.; Kucera, R.; Kripnerova, M.; Srbecka, K.; Hora, M.; Hes, O.; Klecka, J.; Babuska, V.; Rousarova, M.; et al. Prognostic Significance of TMPRSS2-ERG Fusion Gene in Prostate Cancer. Anticancer Res. 2016, 36, 4787–4793. [Google Scholar] [CrossRef] [PubMed]
- Boysen, G.; Barbieri, C.E.; Prandi, D.; Blattner, M.; Chae, S.S.; Dahija, A.; Nataraj, S.; Huang, D.; Marotz, C.; Xu, L.; et al. SPOP mutation leads to genomic instability in prostate cancer. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.J.; Zhao, J.C.; Ogden, I.; Bergan, R.C.; Yu, J. Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer Res. 2013, 73, 3725–3736. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Yu, J. Current perspectives on FOXA1 regulation of androgen receptor signaling and prostate cancer. Genes Dis. 2015, 2, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prensner, J.R.; Chinnaiyan, A.M. Metabolism unhinged: IDH mutations in cancer. Nat. Med. 2011, 17, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.E.; Bittinger, M.A.; Su, S.M.; Fantin, V.R. Cancer-associated IDH mutations: Biomarker and therapeutic opportunities. Oncogene 2010, 29, 6409–6417. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.J.; Shih, H.A.; Andronesi, O.C.; Cahill, D.P. Isocitrate dehydrogenase-mutant glioma: Evolving clinical and therapeutic implications. Cancer 2017, 123, 4535–4546. [Google Scholar] [CrossRef] [PubMed]
- Ghiam, A.F.; Cairns, R.A.; Thoms, J.; Dal Pra, A.; Ahmed, O.; Meng, A.; Mak, T.W.; Bristow, R.G. IDH mutation status in prostate cancer. Oncogene 2012, 31, 3826. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Bangma, C.H.; Bjartell, A.; Catto, J.W.; Culig, Z.; Gronberg, H.; Luo, J.; Visakorpi, T.; Rubin, M.A. The mutational landscape of prostate cancer. Eur. Urol. 2013, 64, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, S.E.; Rao, A.D.; Lyle, M.; Sandhu, S.; Long, G.V.; McArthur, G.A.; Raleigh, J.M.; Hicks, R.J.; Millward, M. Activity of trametinib in K601E and L597Q BRAF mutation-positive metastatic melanoma. Melanoma Res. 2014, 24, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Boysen, G.; Barbieri, C.E.; Bryant, H.E.; Castro, E.; Nelson, P.S.; Olmos, D.; Pritchard, C.C.; Rubin, M.A.; de Bono, J.S. DNA Repair in Prostate Cancer: Biology and Clinical Implications. Eur. Urol. 2017, 71, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Weight, C.J.; Narayan, V.M.; Smith, D.; Kim, S.P.; Karnes, R.J. The Effects of Population-based Prostate-specific Antigen Screening Beginning at Age 40. Urology 2017, 110, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Grossman, D.C.; Curry, S.J.; Owens, D.K.; Bibbins-Domingo, K.; Caughey, A.B.; Davidson, K.W.; Doubeni, C.A.; Ebell, M.; Epling, J.W., Jr.; Kemper, A.R.; et al. Screening for Prostate Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 319, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Sathianathen, N.J.; Konety, B.R.; Crook, J.; Saad, F.; Lawrentschuk, N. Landmarks in prostate cancer. Nat. Rev. Urol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mottet, N.; Bellmunt, J.; Bolla, M.; Briers, E.; Cumberbatch, M.G.; De Santis, M.; Fossati, N.; Gross, T.; Henry, A.M.; Joniau, S.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2017, 71, 618–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussemakers, M.J.; van Bokhoven, A.; Verhaegh, G.W.; Smit, F.P.; Karthaus, H.F.; Schalken, J.A.; Debruyne, F.M.; Ru, N.; Isaacs, W.B. DD3: A new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 1999, 59, 5975–5979. [Google Scholar] [PubMed]
- Wei, J.T.; Feng, Z.; Partin, A.W.; Brown, E.; Thompson, I.; Sokoll, L.; Chan, D.W.; Lotan, Y.; Kibel, A.S.; Busby, J.E.; et al. Can urinary PCA3 supplement PSA in the early detection of prostate cancer? J. Clin. Oncol. 2014, 32, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Salami, S.S.; Schmidt, F.; Laxman, B.; Regan, M.M.; Rickman, D.S.; Scherr, D.; Bueti, G.; Siddiqui, J.; Tomlins, S.A.; Wei, J.T.; et al. Combining urinary detection of TMPRSS2:ERG and PCA3 with serum PSA to predict diagnosis of prostate cancer. Urol. Oncol. 2013, 31, 566–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Neste, L.; Hendriks, R.J.; Dijkstra, S.; Trooskens, G.; Cornel, E.B.; Jannink, S.A.; de Jong, H.; Hessels, D.; Smit, F.P.; Melchers, W.J.; et al. Detection of High-grade Prostate Cancer Using a Urinary Molecular Biomarker-Based Risk Score. Eur. Urol. 2016, 70, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Zappala, S.M.; Scardino, P.T.; Okrongly, D.; Linder, V.; Dong, Y. Clinical performance of the 4Kscore Test to predict high-grade prostate cancer at biopsy: A meta-analysis of us and European clinical validation study results. Rev. Urol. 2017, 19, 149–155. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Shenoy, B.V.; Tutrone, R.F.; Karsh, L.I.; Saltzstein, D.R.; Harmon, W.J.; Broyles, D.L.; Roddy, T.E.; Lofaro, L.R.; Paoli, C.J.; et al. Clinical utility of the Prostate Health Index (phi) for biopsy decision management in a large group urology practice setting. Prostate Cancer Prostatic Dis. 2018, 21, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Van Neste, L.; Herman, J.G.; Otto, G.; Bigley, J.W.; Epstein, J.I.; Van Criekinge, W. The epigenetic promise for prostate cancer diagnosis. Prostate 2012, 72, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, J.; Varde, S.; Wang, H.; Chiu, H.; Vargo, J.; Gray, K.; Nagle, R.B.; Neri, J.R.; Mazumder, A. Quantitative, spatial resolution of the epigenetic field effect in prostate cancer. Prostate 2008, 68, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.R.; Parsons, J.K.; Andriole, G.; Bahnson, R.R.; Castle, E.P.; Catalona, W.J.; Dahl, D.M.; Davis, J.W.; Epstein, J.I.; Etzioni, R.B.; et al. NCCN Guidelines Insights: Prostate Cancer Early Detection, Version 2.2016. J. Natl. Compr. Canc. Netw. 2016, 14, 509–519. [Google Scholar] [CrossRef] [PubMed]
- NCCN Guidelines Version 2.2018 Prostate Cancer Early Detection. Available online: https://www.nccn.org/professionals/physician_gls/PDF/prostate_detection.pdf (accessed on 27 July 2018).
- McDermed, J.E.; Sanders, R.; Fait, S.; Klem, R.E.; Sarno, M.J.; Adams, T.H.; Diamandis, E.P. Nucleic acid detection immunoassay for prostate-specific antigen based on immuno-PCR methodology. Clin. Chem. 2012, 58, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Moul, J.W.; Lilja, H.; Semmes, O.J.; Lance, R.S.; Vessella, R.L.; Fleisher, M.; Mazzola, C.; Sarno, M.J.; Stevens, B.; Klem, R.E.; et al. NADiA ProsVue prostate-specific antigen slope is an independent prognostic marker for identifying men at reduced risk of clinical recurrence of prostate cancer after radical prostatectomy. Urology 2012, 80, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Moul, J.W.; Sarno, M.J.; McDermed, J.E.; Triebell, M.T.; Reynolds, M.A. NADiA ProsVue prostate-specific antigen slope, CAPRA-S, and prostate cancer--specific survival after radical prostatectomy. Urology 2014, 84, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.H.; Mohler, J.L. NCCN Guidelines Updates: Prostate Cancer and Prostate Cancer Early Detection. J. Natl. Compr. Canc. Netw. 2018, 16, 620–623. [Google Scholar] [CrossRef] [PubMed]
- NCCN Guidelines Version 2.2018 Prostate Cancer. Available online: https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf (accessed on 27 July 2018).
- Erho, N.; Crisan, A.; Vergara, I.A.; Mitra, A.P.; Ghadessi, M.; Buerki, C.; Bergstralh, E.J.; Kollmeyer, T.; Fink, S.; Haddad, Z.; et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS ONE 2013, 8, e66855. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.; Rosner, I.L.; Brand, T.C.; Zhang, N.; Tsiatis, A.C.; Moncur, J.; Ali, A.; Chen, Y.; Knezevic, D.; Maddala, T.; et al. A Biopsy-based 17-gene Genomic Prostate Score Predicts Recurrence After Radical Prostatectomy and Adverse Surgical Pathology in a Racially Diverse Population of Men with Clinically Low- and Intermediate-risk Prostate Cancer. Eur. Urol. 2015, 68, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, E.A.; Cooperberg, M.R.; Magi-Galluzzi, C.; Simko, J.P.; Falzarano, S.M.; Maddala, T.; Chan, J.M.; Li, J.; Cowan, J.E.; Tsiatis, A.C.; et al. A 17-gene assay to predict prostate cancer aggressiveness in the context of Gleason grade heterogeneity, tumor multifocality, and biopsy undersampling. Eur. Urol. 2014, 66, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef]
- Sommariva, S.; Tarricone, R.; Lazzeri, M.; Ricciardi, W.; Montorsi, F. Prognostic Value of the Cell Cycle Progression Score in Patients with Prostate Cancer: A Systematic Review and Meta-analysis. Eur. Urol. 2016, 69, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Shipitsin, M.; Small, C.; Choudhury, S.; Giladi, E.; Friedlander, S.; Nardone, J.; Hussain, S.; Hurley, A.D.; Ernst, C.; Huang, Y.E.; et al. Identification of proteomic biomarkers predicting prostate cancer aggressiveness and lethality despite biopsy-sampling error. Br. J. Cancer 2014, 111, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Shipitsin, M.; Small, C.; Giladi, E.; Siddiqui, S.; Choudhury, S.; Hussain, S.; Huang, Y.E.; Chang, H.; Rimm, D.L.; Berman, D.M.; et al. Automated quantitative multiplex immunofluorescence in situ imaging identifies phospho-S6 and phospho-PRAS40 as predictive protein biomarkers for prostate cancer lethality. Proteome Sci. 2014, 12, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlins, S.A.; Day, J.R.; Lonigro, R.J.; Hovelson, D.H.; Siddiqui, J.; Kunju, L.P.; Dunn, R.L.; Meyer, S.; Hodge, P.; Groskopf, J.; et al. Urine TMPRSS2:ERG Plus PCA3 for Individualized Prostate Cancer Risk Assessment. Eur. Urol. 2016, 70, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanda, M.G.; Feng, Z.; Howard, D.H.; Tomlins, S.A.; Sokoll, L.J.; Chan, D.W.; Regan, M.M.; Groskopf, J.; Chipman, J.; Patil, D.H.; et al. Association Between Combined TMPRSS2:ERG and PCA3 RNA Urinary Testing and Detection of Aggressive Prostate Cancer. JAMA Oncol. 2017, 3, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Parekh, D.J.; Punnen, S.; Sjoberg, D.D.; Asroff, S.W.; Bailen, J.L.; Cochran, J.S.; Concepcion, R.; David, R.D.; Deck, K.B.; Dumbadze, I.; et al. A multi-institutional prospective trial in the USA confirms that the 4Kscore accurately identifies men with high-grade prostate cancer. Eur. Urol. 2015, 68, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Punnen, S.; Freedland, S.J.; Polascik, T.J.; Loeb, S.; Risk, M.C.; Savage, S.; Mathur, S.C.; Uchio, E.; Dong, Y.; Silberstein, J.L. A Multi-Institutional Prospective Trial Confirms Noninvasive Blood Test Maintains Predictive Value in African American Men. J. Urol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Roobol, M.J.; Savage, C.J.; Peltola, M.; Pettersson, K.; Scardino, P.T.; Vickers, A.J.; Schroder, F.H.; Lilja, H. A four-kallikrein panel for the prediction of repeat prostate biopsy: Data from the European Randomized Study of Prostate Cancer screening in Rotterdam, Netherlands. Br. J. Cancer 2010, 103, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Partin, A.W.; Van Neste, L.; Klein, E.A.; Marks, L.S.; Gee, J.R.; Troyer, D.A.; Rieger-Christ, K.; Jones, J.S.; Magi-Galluzzi, C.; Mangold, L.A.; et al. Clinical validation of an epigenetic assay to predict negative histopathological results in repeat prostate biopsies. J. Urol. 2014, 192, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.D.; Van Neste, L.; Delvenne, P.; Delree, P.; Delga, A.; McNeill, S.A.; O’Donnell, M.; Clark, J.; Van Criekinge, W.; Bigley, J.; et al. Clinical utility of an epigenetic assay to detect occult prostate cancer in histopathologically negative biopsies: Results of the MATLOC study. J. Urol. 2013, 189, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Tosoian, J.J.; Druskin, S.C.; Andreas, D.; Mullane, P.; Chappidi, M.; Joo, S.; Ghabili, K.; Agostino, J.; Macura, K.J.; Carter, H.B.; et al. Use of the Prostate Health Index for detection of prostate cancer: Results from a large academic practice. Prostate Cancer Prostatic Dis. 2017, 20, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Scattoni, V.; Lazzeri, M.; Lughezzani, G.; De Luca, S.; Passera, R.; Bollito, E.; Randone, D.; Abdollah, F.; Capitanio, U.; Larcher, A.; et al. Head-to-head comparison of prostate health index and urinary PCA3 for predicting cancer at initial or repeat biopsy. J. Urol. 2013, 190, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Clinton, T.N.; Bagrodia, A.; Lotan, Y.; Margulis, V.; Raj, G.V.; Woldu, S.L. Tissue-based biomarkers in prostate cancer. Expert Rev. Precis. Med. Drug Dev. 2017, 2, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Berman, D.M.; Rimm, D.L.; Shipitsin, M.; Putzi, M.; Nifong, T.P.; Small, C.; Choudhury, S.; Capela, T.; Coupal, L.; et al. Development and clinical validation of an in situ biopsy-based multimarker assay for risk stratification in prostate cancer. Clin. Cancer Res. 2015, 21, 2591–2600. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Zargar-Shoshtari, K.; Pow-Sang, J.M. Biomarkers for prostate cancer: Present challenges and future opportunities. Future Sci. OA 2016, 2, FSO72. [Google Scholar] [CrossRef] [PubMed]
- Strand, S.H.; Orntoft, T.F.; Sorensen, K.D. Prognostic DNA methylation markers for prostate cancer. Int. J. Mol. Sci. 2014, 15, 16544–16576. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J.L.; Armstrong, A.J.; Bahnson, R.R.; D’Amico, A.V.; Davis, B.J.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; Horwitz, E.M.; et al. Prostate Cancer, Version 1.2016. J. Natl. Compr. Canc. Netw. 2016, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Bill-Axelson, A.; Holmberg, L.; Filen, F.; Ruutu, M.; Garmo, H.; Busch, C.; Nordling, S.; Haggman, M.; Andersson, S.O.; Bratell, S.; et al. Radical prostatectomy versus watchful waiting in localized prostate cancer: The Scandinavian prostate cancer group-4 randomized trial. J. Natl. Cancer Inst. 2008, 100, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Bill-Axelson, A.; Holmberg, L.; Garmo, H.; Rider, J.R.; Taari, K.; Busch, C.; Nordling, S.; Haggman, M.; Andersson, S.O.; Spangberg, A.; et al. Radical prostatectomy or watchful waiting in early prostate cancer. N. Engl. J. Med. 2014, 370, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.U.; Hindley, R.G.; Dickinson, L.; Freeman, A.; Kirkham, A.P.; Sahu, M.; Scott, R.; Allen, C.; Van der Meulen, J.; Emberton, M. Focal therapy for localised unifocal and multifocal prostate cancer: A prospective development study. Lancet Oncol. 2012, 13, 622–632. [Google Scholar] [CrossRef]
- Messing, E.M.; Manola, J.; Yao, J.; Kiernan, M.; Crawford, D.; Wilding, G.; di’SantAgnese, P.A.; Trump, D. On behalf of the Eastern Cooperative Oncology Group study EST 3886. Immediate versus deferred androgen deprivation treatment in patients with node-positive prostate cancer after radical prostatectomy and pelvic lymphadenectomy. Lancet Oncol. 2006, 7, 472–479. [Google Scholar] [CrossRef]
- Safety and Efficacy Study of Enzalutamide Plus Leuprolide in Patients With Nonmetastatic Prostate Cancer (EMBARK). Available online: https://ClinicalTrials.gov/show/NCT02319837 (accessed on 27 July 2018).
- Bubendorf, L.; Schopfer, A.; Wagner, U.; Sauter, G.; Moch, H.; Willi, N.; Gasser, T.C.; Mihatsch, M.J. Metastatic patterns of prostate cancer: An autopsy study of 1589 patients. Hum. Pathol. 2000, 31, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; de Bono, J.S. Metastatic Prostate Cancer. N. Engl. J. Med. 2018, 378, 1653–1654. [Google Scholar] [CrossRef] [PubMed]
- Montero, A.; Fossella, F.; Hortobagyi, G.; Valero, V. Docetaxel for treatment of solid tumours: A systematic review of clinical data. Lancet Oncol. 2005, 6, 229–239. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; McKiernan, J.; Eastham, J. Rationale for zoledronic acid therapy in men with hormone-sensitive prostate cancer with or without bone metastasis. Urol. Oncol. 2006, 24, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Carducci, M.; Smith, M.; Damiao, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef]
- Brown, J.E.; Cleeland, C.S.; Fallowfield, L.J.; Patrick, D.L.; Fizazi, K.; Smith, M.R.; Maroto, J.P.; Michel, M.S.; Feng, A.; Goessl, C.; et al. Pain outcomes in patients with bone metastases from castrate-resistant prostate cancer: Results from a phase 3 trials of denosumab vs. zoledronic acid. Eur. Urol. Suppl. 2011, 10, 336. [Google Scholar] [CrossRef]
- Patrick, D.; Cleeland, C.S.; Fallowfield, L.; Smith, M.R.; Klotz, L.; Oudard, S.; Marx, G.M.; Wei, R.; Ohrling, K.; Qian, Y. Denosumab or zoledronic acid (ZA) therapy on pain interference and cancer-specific quality of life (CSQoL) in patients with castrate-resistant prostate cancer (CRPC) and bone metastases (BM). J. Clin. Oncol. 2014, 32, 12. [Google Scholar] [CrossRef]
- Hegemann, M.; Bedke, J.; Stenzl, A.; Todenhofer, T. Denosumab treatment in the management of patients with advanced prostate cancer: Clinical evidence and experience. Ther. Adv. Urol. 2017, 9, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Takeda, D.Y.; Spisak, S.; Seo, J.H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szallasi, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Xia, J.-H.; Sipeky, C.; Dong, X.-M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and Clinical Implications of the 19q13 Aggressive Prostate Cancer Susceptibility Locus. Cell 2018, 174, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.; Shibata, Y.; Hall, I.M.; Dutta, A. Chromosomal structural variations during progression of a prostate epithelial cell line to a malignant metastatic state inactivate the NF2, NIPSNAP1, UGT2B17, and LPIN2 genes. Cancer Biol. Ther. 2013, 14, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Jaratlerdsiri, W.; Chan, E.K.F.; Petersen, D.C.; Yang, C.; Croucher, P.I.; Bornman, M.S.R.; Sheth, P.; Hayes, V.M. Next generation mapping reveals novel large genomic rearrangements in prostate cancer. Oncotarget 2017, 8, 23588–23602. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M.; Cunha, I.W.; Coudry, R.A.; Fonseca, F.P.; Torres, C.H.; Soares, F.A.; Squire, J.A. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br. J. Cancer 2007, 97, 678–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, L.M.; Miller, T.W. Therapeutic targeting of cancers with loss of PTEN function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; De Giorgi, U.; Massard, C.; Bracarda, S.; Nava Rodrigues, D.; Kocak, I.; Font, A.; Arija, J.A.; Shih, K.; Radavoi, G.D.; et al. PTEN loss as a predictive biomarker for the Akt inhibitor ipatasertib combined with abiraterone acetate in patients with metastatic castration-resistant prostate cancer (mCRPC). Ann. Oncol. 2016, 27, 718O. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Chen, Y.H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.R.; Yu, M.K.; Small, E.J. Apalutamide and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 2542. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Z.; Xiao, W.; Yan, L.; Guan, W.; Hu, Z.; Wu, L.; Huang, Q.; Wang, J.; Xu, H.; et al. Androgen-receptor splice variant-7-positive prostate cancer: A novel molecular subtype with markedly worse androgen-deprivation therapy outcomes in newly diagnosed patients. Mod. Pathol. 2018, 31, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Papazoglou, D.; Wannesson, L.; Berthold, D.; Cathomas, R.; Gillessen, S.; Rothermundt, C.; Hasler, L.; Winterhalder, R.; Barth, A.; Mingrone, W.; et al. Enzalutamide in Patients With Castration-Resistant Prostate Cancer Progressing After Docetaxel: Retrospective Analysis of the Swiss Enzalutamide Named Patient Program. Clin. Genitourin Cancer 2017, 15, e315–e323. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.; Tombolini, V.; Marampon, F.; Musella, A.; Marchetti, C. Defective DNA repair mechanisms in prostate cancer: Impact of olaparib. Drug Des. Devel. Ther. 2017, 11, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ph II Study to Evaluate Olaparib With Abiraterone in Treating Metastatic Castration Resistant Prostate Cancer. Available online: https://ClinicalTrials.gov/show/NCT01972217 (accessed on 27 July 2018).
- Phase I/II Study of the Anti-Programmed Death Ligand-1 Antibody MEDI4736 in Combination With Olaparib and/or Cediranib for Advanced Solid Tumors and Advanced or Recurrent Ovarian, Triple Negative Breast, Lung, Prostate and Colorectal Cancers. Available online: https://ClinicalTrials.gov/show/NCT02484404 (accessed on 27 July 2018).
- TOPARP: A Phase II Trial of Olaparib in Patients With Advanced Castration Resistant Prostate Cancer. Available online: https://ClinicalTrials.gov/show/NCT01682772 (accessed on 27 July 2018).
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Brenner, J.C.; Ateeq, B.; Li, Y.; Yocum, A.K.; Cao, Q.; Asangani, I.A.; Patel, S.; Wang, X.; Liang, H.; Yu, J.; et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell 2011, 19, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Daignault-Newton, S.; Twardowski, P.W.; Albany, C.; Stein, M.N.; Kunju, L.P.; Siddiqui, J.; Wu, Y.M.; Robinson, D.; Lonigro, R.J.; et al. Targeting Androgen Receptor and DNA Repair in Metastatic Castration-Resistant Prostate Cancer: Results From NCI 9012. J. Clin. Oncol. 2018, 36, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Choudhary, G.S.; Sharma, A.; Singh, K.; Heston, W.D.; Ciezki, J.; Klein, E.A.; Almasan, A. PARP inhibition sensitizes to low dose-rate radiation TMPRSS2-ERG fusion gene-expressing and PTEN-deficient prostate cancer cells. PLoS ONE 2013, 8, e60408. [Google Scholar] [CrossRef] [PubMed]
- Buonerba, C.; Federico, P.; D’Aniello, C.; Rescigno, P.; Cavaliere, C.; Puglia, L.; Ferro, M.; Altieri, V.; Perdona, S.; De Placido, S.; et al. Phase II trial of cisplatin plus prednisone in docetaxel-refractory castration-resistant prostate cancer patients. Cancer Chemother. Pharmacol. 2011, 67, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Kentepozidis, N.; Soultati, A.; Giassas, S.; Vardakis, N.; Kalykaki, A.; Kotsakis, A.; Papadimitraki, E.; Pantazopoulos, N.; Bozionellou, V.; Georgoulias, V. Paclitaxel in combination with carboplatin as salvage treatment in patients with castration-resistant prostate cancer: A Hellenic oncology research group multicenter phase II study. Cancer Chemother. Pharmacol. 2012, 70, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Urakami, S.; Igawa, M.; Kikuno, N.; Yoshino, T.; Kishi, H.; Shigeno, K.; Shiina, H. Combination chemotherapy with paclitaxel, estramustine and carboplatin for hormone refractory prostate cancer. J. Urol. 2002, 168, 2444–2450. [Google Scholar] [CrossRef]
- Droz, J.P.; Muracciole, X.; Mottet, N.; Ould Kaci, M.; Vannetzel, J.M.; Albin, N.; Culine, S.; Rodier, J.M.; Misset, J.L.; Mackenzie, S.; et al. Phase II study of oxaliplatin versus oxaliplatin combined with infusional 5-fluorouracil in hormone refractory metastatic prostate cancer patients. Ann. Oncol. 2003, 14, 1291–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culine, S.; El Demery, M.; Lamy, P.J.; Iborra, F.; Avances, C.; Pinguet, F. Docetaxel and cisplatin in patients with metastatic androgen independent prostate cancer and circulating neuroendocrine markers. J. Urol. 2007, 178, 844–848, discussion 848. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, C.N.; Daliani, D.D.; Thall, P.F.; Tu, S.M.; Wang, X.; Reyes, A.; Troncoso, P.; Logothetis, C.J. Results of a phase II study with doxorubicin, etoposide, and cisplatin in patients with fully characterized small-cell carcinoma of the prostate. J. Clin. Oncol. 2002, 20, 3072–3080. [Google Scholar] [CrossRef] [PubMed]
- Miglietta, L.; Cannobbio, L.; Boccardo, F. Assessment of response to carboplatin in patients with hormone-refractory prostate cancer: A critical analysis of drug activity. Anticancer Res. 1995, 15, 2825–2828. [Google Scholar] [PubMed]
- Sternberg, C.N.; Petrylak, D.P.; Sartor, O.; Witjes, J.A.; Demkow, T.; Ferrero, J.M.; Eymard, J.C.; Falcon, S.; Calabro, F.; James, N.; et al. Multinational, double-blind, phase III study of prednisone and either satraplatin or placebo in patients with castrate-refractory prostate cancer progressing after prior chemotherapy: The SPARC trial. J. Clin. Oncol. 2009, 27, 5431–5438. [Google Scholar] [CrossRef] [PubMed]
- Hager, S.; Ackermann, C.J.; Joerger, M.; Gillessen, S.; Omlin, A. Anti-tumour activity of platinum compounds in advanced prostate cancer-a systematic literature review. Ann. Oncol. 2016, 27, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Carboplatin in Castration-resistant Prostate Cancer. Available online: https://ClinicalTrials.gov/show/NCT02311764 (accessed on 20 August 2018).
- Docetaxel and Carboplatin in Treating Patients With Metastatic, Castration Resistant Prostate Cancer Containing Inactivated Genes in the BRCA 1/2 Pathway. Available online: https://ClinicalTrials.gov/show/NCT02598895 (accessed on 20 August 2018).
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Morrissey, C.; Kumar, A.; Zhang, X.; Smith, C.; Coleman, I.; Salipante, S.J.; Milbank, J.; Yu, M.; Grady, W.M.; et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat. Commun. 2014, 5, 4988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu-Monette, Z.Y.; Zhang, M.; Li, J.; Young, K.H. PD-1/PD-L1 Blockade: Have We Found the Key to Unleash the Antitumor Immune Response? Front. Immunol. 2017, 8, 1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graff, J.N.; Alumkal, J.J.; Drake, C.G.; Thomas, G.V.; Redmond, W.L.; Farhad, M.; Cetnar, J.P.; Ey, F.S.; Bergan, R.C.; Slottke, R.; et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget 2016, 7, 52810–52817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, A.; Massard, C.; Ott, P.A.; Haas, N.; Lopez, J.; Ejadi, S.; Wallmark, J.; Keam, B.; Delord, J.P.; Aggarwal, R.; et al. Pembrolizumab for patients with advanced prostate adenocarcinoma: Preliminary results from the KEYNOTE-028 study. Ann. Oncol. 2016, 27, 725PD. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.L.; Sio, A.; Angeles, A.; Roberts, M.E.; Azad, A.A.; Chi, K.N.; Zoubeidi, A. PD-L1 is highly expressed in Enzalutamide resistant prostate cancer. Oncotarget 2015, 6, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Guner, G.; Taheri, D.; Netto, G.J.; Palsgrove, D.N.; Zheng, Q.; Guedes, L.B.; Kim, K.; Tsai, H.; Esopi, D.M.; et al. Comprehensive Evaluation of Programmed Death-Ligand 1 Expression in Primary and Metastatic Prostate Cancer. Am. J. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Cieslik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782 e1714. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Papavassiliou, K.A.; Papavassiliou, A.G. Bromodomains: Pockets with therapeutic potential. Trends Mol. Med. 2014, 20, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, T.; Schultz, D.; Hicks, J.; Hubbard, G.K.; Mutton, L.N.; Lotan, T.L.; Bethel, C.; Lotz, M.T.; Yegnasubramanian, S.; Nelson, W.G.; et al. MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PLoS ONE 2010, 5, e9427. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.B.; Qian, J.; Lieber, M.M.; Bostwick, D.G. Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res. 1997, 57, 524–531. [Google Scholar] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asangani, I.A.; Wilder-Romans, K.; Dommeti, V.L.; Krishnamurthy, P.M.; Apel, I.J.; Escara-Wilke, J.; Plymate, S.R.; Navone, N.M.; Wang, S.; Feng, F.Y.; et al. BET Bromodomain Inhibitors Enhance Efficacy and Disrupt Resistance to AR Antagonists in the Treatment of Prostate Cancer. Mol. Cancer Res. 2016, 14, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Gan, W.; Li, X.; Wang, S.; Zhang, W.; Huang, L.; Liu, S.; Zhong, Q.; Guo, J.; Zhang, J.; et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 2017, 23, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, D.; Zhao, Y.; Ren, S.; Gao, K.; Ye, Z.; Wang, S.; Pan, C.W.; Zhu, Y.; Yan, Y.; et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med. 2017, 23, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- A Study of ZEN003694 in Patients With Metastatic Castration-Resistant Prostate Cancer. Available online: https://ClinicalTrials.gov/show/NCT02705469 (accessed on 27 July 2018).
- A Dose-Finding Study of OTX105/MK-8628, a Small Molecule Inhibitor of the Bromodomain and Extra-Terminal (BET) Proteins, in Adults With Selected Advanced Solid Tumors (MK-8628-003). Available online: https://ClinicalTrials.gov/show/NCT02259114 (accessed on 27 July 2018).
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, J.I.; Amin, M.B.; Beltran, H.; Lotan, T.L.; Mosquera, J.M.; Reuter, V.E.; Robinson, B.D.; Troncoso, P.; Rubin, M.A. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Pellecchia, M.; Ronai, Z.A. The Siah2-HIF-FoxA2 axis in prostate cancer—new markers and therapeutic opportunities. Oncotarget 2010, 1, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.T.; Yao, Y.H.; Li, B.G.; Tang, Y.; Chang, J.W.; Zhang, J. Neuroendocrine Prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: Factors associated with time to development of NEPC and survival from NEPC diagnosis-a systematic review and pooled analysis. J. Clin. Oncol. 2014, 32, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Gillessen, S.; Omlin, A.; Attard, G.; de Bono, J.S.; Efstathiou, E.; Fizazi, K.; Halabi, S.; Nelson, P.S.; Sartor, O.; Smith, M.R.; et al. Management of patients with advanced prostate cancer: Recommendations of the St Gallen Advanced Prostate Cancer Consensus Conference (APCCC) 2015. Ann. Oncol. 2015, 26, 1589–1604. [Google Scholar] [CrossRef] [PubMed]
- Bonkhoff, H.; Stein, U.; Remberger, K. Endocrine-paracrine cell types in the prostate and prostatic adenocarcinoma are postmitotic cells. Hum. Pathol. 1995, 26, 167–170. [Google Scholar] [CrossRef]
- Sauer, C.G.; Roemer, A.; Grobholz, R. Genetic analysis of neuroendocrine tumor cells in prostatic carcinoma. Prostate 2006, 66, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Gupta, N.S.; Wang, W.; Toubaji, A.; Haffner, M.C.; Chaux, A.; Hicks, J.L.; Meeker, A.K.; Bieberich, C.J.; De Marzo, A.M.; et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod. Pathol. 2011, 24, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel, D.E.; Nakayama, M.; Luo, J.; Abukhdeir, A.M.; Park, B.H.; Bieberich, C.J.; Hicks, J.L.; Eisenberger, M.; Nelson, W.G.; Mostwin, J.L.; et al. Shared TP53 gene mutation in morphologically and phenotypically distinct concurrent primary small cell neuroendocrine carcinoma and adenocarcinoma of the prostate. Prostate 2009, 69, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Tagawa, S.T.; Park, K.; MacDonald, T.; Milowsky, M.I.; Mosquera, J.M.; Rubin, M.A.; Nanus, D.M. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J. Clin. Oncol. 2012, 30, e386–e389. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Epstein, J.I. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am. J. Surg. Pathol. 2008, 32, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.C.; Veeramani, S.; Lin, F.F.; Kondrikou, D.; Zelivianski, S.; Igawa, T.; Karan, D.; Batra, S.K.; Lin, M.F. Androgen deprivation induces human prostate epithelial neuroendocrine differentiation of androgen-sensitive LNCaP cells. Endocr. Relat. Cancer 2006, 13, 151–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, A.M.; Shen, L.; Tapia, E.L.; Lu, J.F.; Chen, H.C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.; et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016, 22, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- ProSTAR: A Study Evaluating CPI-1205 in Patients With Metastatic Castration Resistant Prostate Cancer. Available online: https://ClinicalTrials.gov/show/NCT03480646 (accessed on 27 July 2018).
- Svensson, C.; Ceder, J.; Iglesias-Gato, D.; Chuan, Y.C.; Pang, S.T.; Bjartell, A.; Martinez, R.M.; Bott, L.; Helczynski, L.; Ulmert, D.; et al. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res. 2014, 42, 999–1015. [Google Scholar] [CrossRef] [PubMed]
- Borno, S.T.; Fischer, A.; Kerick, M.; Falth, M.; Laible, M.; Brase, J.C.; Kuner, R.; Dahl, A.; Grimm, C.; Sayanjali, B.; et al. Genome-wide DNA methylation events in TMPRSS2-ERG fusion-negative prostate cancers implicate an EZH2-dependent mechanism with miR-26a hypermethylation. Cancer Discov. 2012, 2, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Manfredi, M.; Ecsedy, J.A. Scientific Rationale Supporting the Clinical Development Strategy for the Investigational Aurora A Kinase Inhibitor Alisertib in Cancer. Front. Oncol. 2015, 5, 189. [Google Scholar] [CrossRef] [PubMed]
- Kivinummi, K.; Urbanucci, A.; Leinonen, K.; Tammela, T.L.J.; Annala, M.; Isaacs, W.B.; Bova, G.S.; Nykter, M.; Visakorpi, T. The expression of AURKA is androgen regulated in castration-resistant prostate cancer. Sci. Rep. 2017, 7, 17978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potosky, A.L.; Davis, W.W.; Hoffman, R.M.; Stanford, J.L.; Stephenson, R.A.; Penson, D.F.; Harlan, L.C. Five-year outcomes after prostatectomy or radiotherapy for prostate cancer: The prostate cancer outcomes study. J. Natl. Cancer Inst. 2004, 96, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Alisertib, Abiraterone Acetate and Prednisone in Treating Patients With Hormone-Resistant Prostate Cancer. Available online: https://ClinicalTrials.gov/show/NCT01848067 (accessed on 27 July 2018).
- Dash, A.; Chakraborty, S.; Pillai, M.R.; Knapp, F.F., Jr. Peptide receptor radionuclide therapy: An overview. Cancer Biother. Radiopharm. 2015, 30, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Lutetium-177 (Lu177) Prostate-Specific Antigen (PSMA)-Directed EndoRadiotherapy. Available online: https://ClinicalTrials.gov/show/NCT03042312 (accessed on 27 July 2018).
- Penney, K.L.; Stampfer, M.J.; Jahn, J.L.; Sinnott, J.A.; Flavin, R.; Rider, J.R.; Finn, S.; Giovannucci, E.; Sesso, H.D.; Loda, M.; et al. Gleason grade progression is uncommon. Cancer Res. 2013, 73, 5163–5168. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.C.; Hafez, K.S.; Stewart, A.; Montie, J.E.; Wei, J.T. Prostate carcinoma presentation, diagnosis, and staging: An update form the National Cancer Data Base. Cancer 2003, 98, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, I.S. Molecular Testing in Breast Cancer: A Guide to Current Practices. Arch. Pathol. Lab. Med. 2016, 140, 815–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.; Atkins, M.B. Which drug, and when, for patients with BRAF-mutant melanoma? Lancet Oncol. 2013, 14, e60–e69. [Google Scholar] [CrossRef]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.S.; Squire, J.; et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Mol. Diagn. 2013, 15, 415–453. [Google Scholar] [CrossRef] [PubMed]
- Georgi, B.; Korzeniewski, N.; Hadaschik, B.; Grullich, C.; Roth, W.; Sultmann, H.; Pahernik, S.; Hohenfellner, M.; Duensing, S. Evolving therapeutic concepts in prostate cancer based on genome-wide analyses (review). Int. J. Oncol. 2014, 45, 1337–1344. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Test | Company | Sample Input | Testing Platform | Outcome | References |
|---|---|---|---|---|---|
| Clinically available biomarkers for diagnostic assessment | |||||
| ExoDx® | Exosome Diagnostics | Urine | Transcript quantification of PCA3 and TMPRSS2:ERG | Initial biopsy | [38,39] |
| Mi-Prostate Score | University of Michigan | Post-DRE urine | Transcript quantification of PCA3 and TMPRSS2:ERG, in combination with PSA level | Initial biopsy | [82,83] |
| SelectMDx | MDxHealth | Post-DRE urine | Transcript quantification of DLX1 and HOXC6 | Initial biopsy | [63] |
| Progensa PCA3 | Hologic | Post-DRE urine | Transcript quantification of PCA3 and PSA | Rebiopsy | [60,61] |
| 4Kscore® | OPKO Diagnostics | Blood | Immunoassay panel for tPSA, fPSA, iPSA, and hK2 | Rebiopsy | [64,84,85,86] |
| ConfirmMDx | MDxHealth | Prostate tissue | PCR-based determination of the methylation status of GSTP1, APC, and RASSF1 | Rebiopsy | [66,67,87,88] |
| Prostate Health Index | Beckman Coulter | Blood | Immunoassay panel for tPSA, fPSA, and p2PSA | Initial biopsy/rebiopsy | [65,89,90] |
| Molecular panels for prognosis and risk stratification | |||||
| NaDiA® ProsVueTM | Beckman Coulter | Blood | Total serum PSA | Management post-RP | [70,71] |
| DecipherTM | GenomeDx Biosciences | RP | Transcriptomic microarray profiling of 22 gene markers | Management post-RP | [75,91] |
| Oncotype DX® | Genomic Health | Prostate tissue | Quantitative PCR for 12 PCa-related genes and 5 housekeeping controls | Active surveillance or treatment initiation | [76,77] |
| Prolaris® | Myriad Genetics | Prostate tissue | Quantitative PCR for 31 cell cycle-related genes and 15 housekeeping controls | Active surveillance or treatment initiation | [78,79] |
| ProMark® | Metamark | Prostate tissue | In situ measurement of 8 protein biomarkers by automated image analysis | Active surveillance or treatment initiation | [80,81,92] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angeles, A.K.; Bauer, S.; Ratz, L.; Klauck, S.M.; Sültmann, H. Genome-Based Classification and Therapy of Prostate Cancer. Diagnostics 2018, 8, 62. https://doi.org/10.3390/diagnostics8030062
Angeles AK, Bauer S, Ratz L, Klauck SM, Sültmann H. Genome-Based Classification and Therapy of Prostate Cancer. Diagnostics. 2018; 8(3):62. https://doi.org/10.3390/diagnostics8030062
Chicago/Turabian StyleAngeles, Arlou Kristina, Simone Bauer, Leonie Ratz, Sabine M. Klauck, and Holger Sültmann. 2018. "Genome-Based Classification and Therapy of Prostate Cancer" Diagnostics 8, no. 3: 62. https://doi.org/10.3390/diagnostics8030062
APA StyleAngeles, A. K., Bauer, S., Ratz, L., Klauck, S. M., & Sültmann, H. (2018). Genome-Based Classification and Therapy of Prostate Cancer. Diagnostics, 8(3), 62. https://doi.org/10.3390/diagnostics8030062