
Drug Repurposing to Treat Glucocorticoid Resistance in Asthma


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Statistical Analysis
2.3. Connectivity Map Analysis
2.4. In Vitro Validation
3. Results
3.1. Demographics
3.2. Differential Gene Expression Analysis
3.3. Connectivity Map Analysis
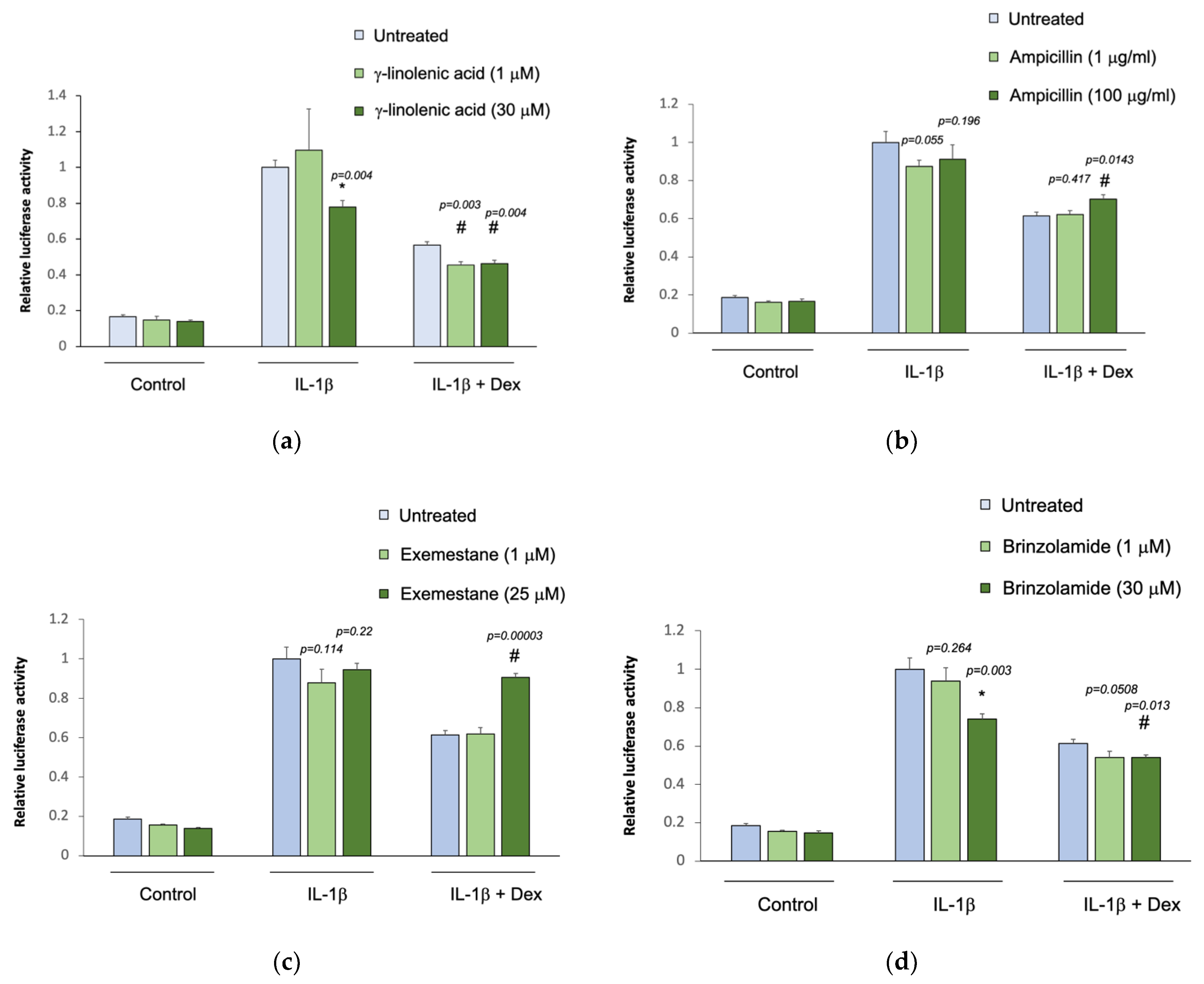
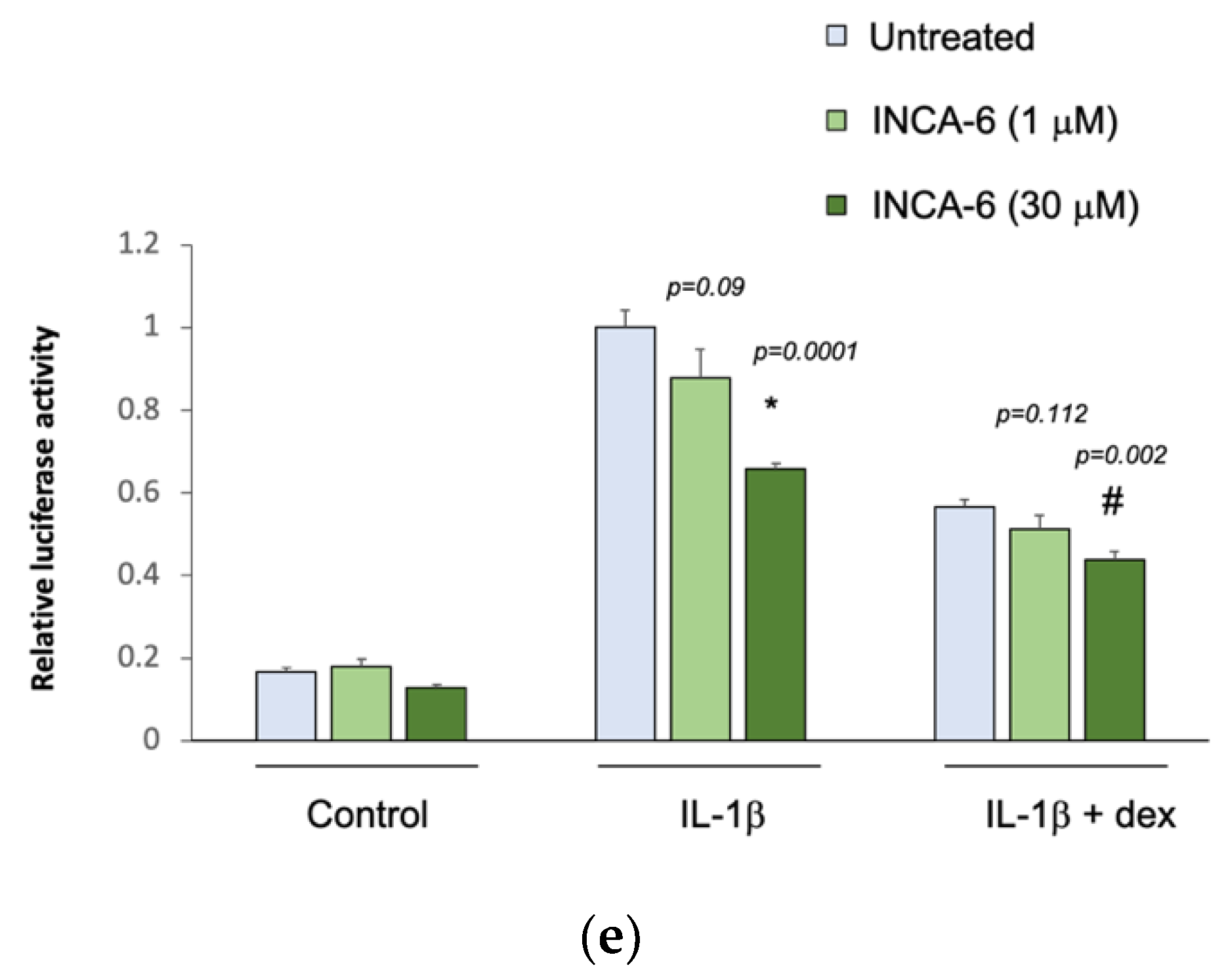
3.4. In Vitro Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]
- Szefler, S.J.; Martin, R.J.; King, T.S.; Boushey, H.A.; Cherniack, R.M.; Chinchilli, V.M.; Craig, T.J.; Dolovich, M.; Drazen, J.M.; Fagan, J.K.; et al. Significant variability in response to inhaled corticosteroids for persistent asthma. J. Allergy Clin. Immunol. 2002, 109, 410–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tantisira, K.G.; Lake, S.; Silverman, E.S.; Palmer, L.J.; Lazarus, R.; Silverman, E.K.; Liggett, S.B.; Gelfand, E.W.; Rosenwasser, L.J.; Richter, B.; et al. Corticosteroid pharmacogenetics: Association of sequence variants in CRHR1 with improved lung function in asthmatics treated with inhaled corticosteroids. Hum. Mol. Genet. 2004, 13, 1353–1359. [Google Scholar] [CrossRef] [Green Version]
- Jang, A.S. Steroid response in refractory asthmatics. Korean J. Intern. Med. 2012, 27, 143–148. [Google Scholar] [CrossRef]
- Suzuki, S.; Ogawa, M.; Ohta, S.; Arima, K.; Nunomura, S.; Nanri, Y.; Mitamura, Y.; Yoshihara, T.; Nakamura, Y.; Yamauchi, K.; et al. The potential for repositioning antithyroid agents as antiasthma drugs. J. Allergy Clin. Immunol. 2016, 138, 1458–1461.e1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miner, K.; Labitzke, K.; Liu, B.; Wang, P.; Henckels, K.; Gaida, K.; Elliott, R.; Chen, J.J.; Liu, L.; Leith, A.; et al. Drug Repurposing: The Anthelmintics Niclosamide and Nitazoxanide Are Potent TMEM16A Antagonists That Fully Bronchodilate Airways. Front. Pharm. 2019, 10, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.; Lee, Y.C.; Kim, S.R.; Kim, S.H.; Park, J. Drug Signature-based Finding of Additional Clinical Use of LC28-0126 for Neutrophilic Bronchial Asthma. Sci. Rep. 2015, 5, 17784. [Google Scholar] [CrossRef] [Green Version]
- Al-Khami, A.A.; Ghonim, M.A.; Del Valle, L.; Ibba, S.V.; Zheng, L.; Pyakurel, K.; Okpechi, S.C.; Garay, J.; Wyczechowska, D.; Sanchez-Pino, M.D.; et al. Fuelling the mechanisms of asthma: Increased fatty acid oxidation in inflammatory immune cells may represent a novel therapeutic target. Clin. Exp. Allergy 2017, 47, 1170–1184. [Google Scholar] [CrossRef]
- Howell, C.; Smith, J.R.; Shute, J.K. Targeting matrix metalloproteinase-13 in bronchial epithelial repair. Clin. Exp. Allergy 2018, 48, 1214–1221. [Google Scholar] [CrossRef] [Green Version]
- Keskin, O.; Farzan, N.; Birben, E.; Akel, H.; Karaaslan, C.; Maitland-van der Zee, A.H.; Wechsler, M.E.; Vijverberg, S.J.; Kalayci, O. Genetic associations of the response to inhaled corticosteroids in asthma: A systematic review. Clin. Transl. Allergy 2019, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himes, B.E.; Jiang, X.; Wagner, P.; Hu, R.; Wang, Q.; Klanderman, B.; Whitaker, R.M.; Duan, Q.; Lasky-Su, J.; Nikolos, C.; et al. RNA-Seq transcriptome profiling identifies CRISPLD2 as a glucocorticoid responsive gene that modulates cytokine function in airway smooth muscle cells. PLoS ONE 2014, 9, e99625. [Google Scholar] [CrossRef]
- Qiu, W.; Guo, F.; Glass, K.; Yuan, G.C.; Quackenbush, J.; Zhou, X.; Tantisira, K.G. Differential connectivity of gene regulatory networks distinguishes corticosteroid response in asthma. J. Allergy Clin. Immunol. 2018, 141, 1250–1258. [Google Scholar] [CrossRef] [Green Version]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Austin, D.H. A Congressional Budget Office Study: Research and Development in the Pharmaceutical Industry; No. 2589; Congress of the United States: Washington, DC, USA, 2006; p. 55.
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e1417. [Google Scholar] [CrossRef]
- So, H.C.; Chau, C.K.; Chiu, W.T.; Ho, K.S.; Lo, C.P.; Yim, S.H.; Sham, P.C. Analysis of genome-wide association data highlights candidates for drug repositioning in psychiatry. Nat. Neurosci. 2017, 20, 1342–1349. [Google Scholar] [CrossRef]
- Nygren, P.; Fryknäs, M.; Agerup, B.; Larsson, R. Repositioning of the anthelmintic drug mebendazole for the treatment for colon cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 2133–2140. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.X.; Chen, R.Q.; Hu, D.X.; Xie, X.Q.; Yu, S.B.; Chen, X.Q. Identification of repaglinide as a therapeutic drug for glioblastoma multiforme. Biochem. Biophys. Res. Commun. 2017, 488, 33–39. [Google Scholar] [CrossRef]
- Kwon, O.S.; Lee, H.; Kong, H.J.; Kwon, E.J.; Park, J.E.; Lee, W.; Kang, S.; Kim, M.; Kim, W.; Cha, H.J. Connectivity map-based drug repositioning of bortezomib to reverse the metastatic effect of GALNT14 in lung cancer. Oncogene 2020. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, H.; Xi, Z.; Rogaeva, E. Drug repositioning for diabetes based on ’omics’ data mining. PLoS ONE 2015, 10, e0126082. [Google Scholar] [CrossRef]
- Vargas, D.M.; De Bastiani, M.A.; Zimmer, E.R.; Klamt, F. Alzheimer’s disease master regulators analysis: Search for potential molecular targets and drug repositioning candidates. Alzheimers Res. 2018, 10, 59. [Google Scholar] [CrossRef]
- Childhood Asthma Management Program Research Group. The Childhood Asthma Management Program (CAMP): Design, rationale, and methods. Control Clin. Trials 1999, 20, 91–120. [Google Scholar] [CrossRef]
- Qiu, W.; Rogers, A.J.; Damask, A.; Raby, B.A.; Klanderman, B.J.; Duan, Q.L.; Tyagi, S.; Niu, S.; Anderson, C.; Cahir-Mcfarland, E.; et al. Pharmacogenomics: Novel loci identification via integrating gene differential analysis and eQTL analysis. Hum. Mol. Genet. 2014, 23, 5017–5024. [Google Scholar] [CrossRef] [Green Version]
- Teague, W.G.; Phillips, B.R.; Fahy, J.V.; Wenzel, S.E.; Fitzpatrick, A.M.; Moore, W.C.; Hastie, A.T.; Bleecker, E.R.; Meyers, D.A.; Peters, S.P.; et al. Baseline Features of the Severe Asthma Research Program (SARP III) Cohort: Differences with Age. J. Allergy Clin. Immunol. Pract. 2018, 6, 545–554.e544. [Google Scholar] [CrossRef]
- Phipatanakul, W.; Mauger, D.T.; Sorkness, R.L.; Gaffin, J.M.; Holguin, F.; Woodruff, P.G.; Ly, N.P.; Bacharier, L.B.; Bhakta, N.R.; Moore, W.C.; et al. Effects of Age and Disease Severity on Systemic Corticosteroid Responses in Asthma. Am. J. Respir. Crit. Care Med. 2017, 195, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Guo, B.; Anderson, C.; Klanderman, B.; Carey, V.; Raby, B. iCheck: QC Pipeline and Data Analysis Tools for High-Dimensional Illumina mRNA Expression Data; R Package Version 1.18.0; 2020. [Google Scholar]
- Carlson, M. org.Hs.eg.db: Genome Wide Annotation for Human; R Package Version 3.8.2; 2019. [Google Scholar]
- Iordanidou, M.; Paraskakis, E.; Tavridou, A.; Paschou, P.; Chatzimichael, A.; Manolopoulos, V.G. G894T polymorphism of eNOS gene is a predictor of response to combination of inhaled corticosteroids with long-lasting beta2-agonists in asthmatic children. Pharmacogenomics 2012, 13, 1363–1372. [Google Scholar] [CrossRef]
- Jiang, X.; Dahlin, A.; Weiss, S.T.; Tantisira, K.; Lu, Q. A high-throughput chemical screen identifies novel inhibitors and enhancers of anti-inflammatory functions of the glucocorticoid receptor. Sci. Rep. 2017, 7, 7405. [Google Scholar] [CrossRef] [Green Version]
- O’Byrne, P.; Fabbri, L.M.; Pavord, I.D.; Papi, A.; Petruzzelli, S.; Lange, P. Asthma progression and mortality: The role of inhaled corticosteroids. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef] [Green Version]
- Innes, J.K.; Calder, P.C. Omega-6 fatty acids and inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2018, 132, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Timoszuk, M.; Bielawska, K.; Skrzydlewska, E. Evening Primrose (Oenothera biennis) Biological Activity Dependent on Chemical Composition. Antioxidants 2018, 7, 108. [Google Scholar] [CrossRef] [Green Version]
- Sergeant, S.; Rahbar, E.; Chilton, F.H. Gamma-linolenic acid, Dihommo-gamma linolenic, Eicosanoids and Inflammatory Processes. Eur. J. Pharm. 2016, 785, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Arm, J.P.; Boyce, J.A.; Wang, L.; Chhay, H.; Zahid, M.; Patil, V.; Govindarajulu, U.; Ivester, P.; Weaver, K.L.; Sergeant, S.; et al. Impact of botanical oils on polyunsaturated fatty acid metabolism and leukotriene generation in mild asthmatics. Lipids Health Dis. 2013, 12, 141. [Google Scholar] [CrossRef] [Green Version]
- Surette, M.E.; Koumenis, I.L.; Edens, M.B.; Tramposch, K.M.; Clayton, B.; Bowton, D.; Chilton, F.H. Inhibition of leukotriene biosynthesis by a novel dietary fatty acid formulation in patients with atopic asthma: A randomized, placebo-controlled, parallel-group, prospective trial. Clin. Ther. 2003, 25, 972–979. [Google Scholar] [CrossRef]
- Surette, M.E.; Stull, D.; Lindemann, J. The impact of a medical food containing gammalinolenic and eicosapentaenoic acids on asthma management and the quality of life of adult asthma patients. Curr. Med. Res. Opin. 2008, 24, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, J.; David Pampe, E.; Peterkin, J.J.; Orozco-Cronin, P.; Belofsky, G.; Stull, D. Clinical study of the effects on asthma-related QOL and asthma management of a medical food in adult asthma patients. Curr. Med. Res. Opin. 2009, 25, 2865–2875. [Google Scholar] [CrossRef] [PubMed]
- Teh, A.L.; Pan, H.; Chen, L.; Ong, M.L.; Dogra, S.; Wong, J.; MacIsaac, J.L.; Mah, S.M.; McEwen, L.M.; Saw, S.M.; et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res. 2014, 24, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Henry, E.K.; Sy, C.B.; Inclan-Rico, J.M.; Espinosa, V.; Ghanny, S.S.; Dwyer, D.F.; Soteropoulos, P.; Rivera, A.; Siracusa, M.C. Carbonic anhydrase enzymes regulate mast cell-mediated inflammation. J. Exp. Med. 2016, 213, 1663–1673. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, B.J.; Yeo, C.T.; Chen-Worsdell, Y.M.; Barnes, P.J.; Chung, K.F. Effect of acetazolamide and amiloride against sodium metabisulphite-induced bronchoconstriction in mild asthma. Thorax 1994, 49, 1096–1098. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, W.J.; Rosenberg, M.; Niven, R.W.; Drazen, J.M.; Israel, E. Acetazolamide and furosemide attenuate asthma induced by hyperventilation of cold, dry air. Am. Rev. Respir. Dis. 1992, 146, 1518–1523. [Google Scholar] [CrossRef]
- Spicuzza, L.; Ciancio, N.; Pellegrino, R.; Bellofiore, S.; Polosa, R.; Ricciardolo, F.L.; Brusasco, V.; Di Maria, G.U. The effect of inhaled furosemide and acetazolamide on bronchoconstriction induced by deep inspiration in asthma. Monaldi. Arch. Chest Dis. 2003, 59, 150–154. [Google Scholar] [PubMed]
- Sano, T.; Nakamura, Y.; Matsunaga, Y.; Takahashi, T.; Azuma, M.; Okano, Y.; Shimizu, E.; Ogushi, F.; Sone, S.; Ogura, T. FK506 and cyclosporin A inhibit granulocyte/macrophage colony-stimulating factor production by mononuclear cells in asthma. Eur. Respir. J. 1995, 8, 1473–1478. [Google Scholar] [PubMed]
- Harrison, C.A.; Bastan, R.; Peirce, M.J.; Munday, M.R.; Peachell, P.T. Role of calcineurin in the regulation of human lung mast cell and basophil function by cyclosporine and FK506. Br. J. Pharm. 2007, 150, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, C.J.; Bungre, J.K.; Assoufi, B.; Cooper, A.E.; Seddon, H.; Kay, A.B. Glucocorticoid resistant asthma: T-lymphocyte steroid metabolism and sensitivity to glucocorticoids and immunosuppressive agents. Eur. Respir. J. 1996, 9, 2077–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, A.G.; Kay, A.B.; Barnes, N.C. Trial of cyclosporin in corticosteroid-dependent chronic severe asthma. Lancet 1992, 339, 324–328. [Google Scholar] [CrossRef]
- Lock, S.H.; Kay, A.B.; Barnes, N.C. Double-blind, placebo-controlled study of cyclosporin A as a corticosteroid-sparing agent in corticosteroid-dependent asthma. Am. J. Respir. Crit. Care Med. 1996, 153, 509–514. [Google Scholar] [CrossRef]
- Normansell, R.; Sayer, B.; Waterson, S.; Dennett, E.J.; Del Forno, M.; Dunleavy, A. Antibiotics for exacerbations of asthma. Cochrane Database Syst. Rev. 2018, 6, Cd002741. [Google Scholar] [CrossRef]
- Yung, J.A.; Fuseini, H.; Newcomb, D.C. Hormones, sex, and asthma. Ann. Allergy Asthma. Immunol. 2018, 120, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Foster, K.A.; Oster, C.G.; Mayer, M.M.; Avery, M.L.; Audus, K.L. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp. Cell Res. 1998, 243, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.J.; Tantisira, K.G.; Fuhlbrigge, A.L.; Litonjua, A.A.; Lasky-Su, J.A.; Szefler, S.J.; Strunk, R.C.; Zeiger, R.S.; Weiss, S.T. Predictors of poor response during asthma therapy differ with definition of outcome. Pharmacogenomics 2009, 10, 1231–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| CAMP | SARP | |||||
|---|---|---|---|---|---|---|
| Good Responders (n = 47) | Poor Responders (n = 48) | p-Value | Good Responders (n = 8) | Poor Responders (n = 11) | p-Value | |
| Age (mean ± SD) | 9.0 (±2.2) | 8.5 (±2.1) | 0.3 | 50.9 (±9.1) | 43.3 (±6.6) | 0.05 |
| Sex | 0.61 | 1.00 | ||||
| Female | 25 (53.2%) | 23 (47.9%) | 7 (87.5%) | 9 (81.8%) | ||
| Male | 22 (46.8%) | 25 (52.1%) | 1 (12.5%) | 2 (18.2%) | ||
| Ancestry | 0.6 | 1.00 | ||||
| European | 39 (83.0%) | 42 (87.5%) | 5 (62.5%) | 8 (72.7%) | ||
| African | 5 (10.6%) | 4 (8.3%) | 3 (37.5%) | 3 (27.3%) | ||
| Hispanic | 1 (2.1%) | 2 (4.2%) | - | - | ||
| Other | 2 (4.3%) | 0 (0.0%) | - | - | ||
| Asthma severity | 0.61 | 0.10 | ||||
| Mild | 23 (48.9%) | 21 (43.8%) | 3 (37.5%) | 1 (9.1%) | ||
| Moderate | 24 (51.1%) | 27 (56.2%) | 2 (25.0%) | 1 (9.1%) | ||
| Severe | - | - | 3 (37.5%) | 9 (81.8%) | ||
| Body mass index (mean ± SD) | 18.1 (±2.8) | 18.0 (±2.9) | 0.9 | 34.9 (±8.2) | 35.3 (±8.3) | 0.90 |
| Baseline serum IgE level (kU/L, median ± IQR) | 230.0 (±310.0) | 150.0 (±236.0) | 0.3 | 205.0 (±396.0) | 36.0 (±195.0) | 0.09 |
| Baseline serum eosinophil count (cells/µL, median ± IQR) | 493.0 (±481.0) | 390.0 (±400.0) | 0.1 | 244.0 (±220.0) | 172.0 (±77.0) | 0.08 |
| Baseline PC20 (median ± IQR) | 0.6 (±1.0) | 1.1 (±1.7) | 0.05 | 0.4 (±0.0) | 1.5 (±0.8) | 0.50 |
| Pre-treatment FEV1 % predicted (mean ± SD) | 86.0% (±15.2%) | 99.8% (±14.1%) | <0.001 | 72.5% (±22.5%) | 82.7% (±11.9%) | 0.20 |
| Post-treatment FEV1 % predicted (mean ± SD) | 101.0% (±11.5%) | 100.5% (±15.1%) | 0.8 | 76.5% (±23.6%) | 73.6% (±17.9%) | 0.80 |
| Pre-treatment FEV1/FVC (mean ± SD) | 75.1% (±9.9%) | 80.8% (±7.4%) | 0.002 | 70.4% (±11.0%) | 75.8% (±8.6%) | 0.20 |
| Post-treatment FEV1/FVC (mean ± SD) | 82.0% (±6.5%) | 81.5% (±8.3%) | 0.7 | 71.6% (±9.2%) | 73.0% (±9.2%) | 0.80 |
| Name | Description | CAMP (n = 95) Connectivity Score | SARP (n = 19) Connectivity Score | Weighted Mean Connectivity Score |
|---|---|---|---|---|
| enrofloxacin | Bacterial DNA gyrase inhibitor | −96.51 | −99.79 | −97.06 |
| SB-203186 | Serotonin receptor antagonist | −93.70 | −91.12 | −93.27 |
| γ-linolenic-acid | Anti-inflammatory omega-6 fatty acid | −94.78 | −80.70 | −92.43 |
| dipropyl-5ct (3-(N,N-Dipropylaminoethyl)-1H-indole-5-carboxamide maleate) | Serotonin receptor agonist | −92.65 | −83.03 | −91.05 |
| ampicillin | Bacterial cell wall synthesis inhibitor | −97.60 | −56.59 | −90.76 |
| exemestane | Aromatase inhibitor | −86.59 | −98.24 | −88.53 |
| brinzolamide | Carbonic anhydrase inhibitor | −86.81 | −89.74 | −87.30 |
| INCA-6 | Calcineurin inhibitor | −87.45 | −78.14 | −85.90 |
| SCH-23390 | Dopamine receptor antagonist | −88.17 | −72.69 | −85.59 |
| brazilin | Nitric oxide production inhibitor | −86.00 | −80.72 | −85.12 |
| pyrazinamide | Fatty acid synthase inhibitor | −82.85 | −90.52 | −84.13 |
| vinburnine | Adrenergic receptor antagonist | −79.35 | −99.25 | −82.67 |
| NS-1619 | Calcium channel activator | −79.38 | −98.59 | −82.58 |
| caffeine | Adenosine receptor antagonist | −78.83 | −81.33 | −79.25 |
| masitinib | KIT inhibitor | −79.32 | −76.71 | −78.88 |
| isoflupredone | Glucocorticoid receptor agonist | −82.06 | −62.39 | −78.78 |
| fluocinonide | Glucocorticoid receptor agonist | −80.62 | −66.33 | −78.24 |
| fluoxetine | Selective serotonin reuptake inhibitor | −74.30 | −96.36 | −77.98 |
| quinpirole | Dopamine receptor agonist | −75.20 | −90.40 | −77.73 |
| saquinavir | HIV protease inhibitor | −72.31 | −94.39 | −75.99 |
| retinol | Retinoid receptor ligand | −72.33 | −80.84 | −73.75 |
| doxapram | Potassium channel blocker | −74.67 | −61.12 | −72.41 |
| somatostatin | Somatostatin receptor agonist | −68.97 | −81.93 | −71.13 |
| thalidomide | TNF production inhibitor | −73.74 | −57.45 | −71.02 |
| L-690488 | Inositol monophosphatase inhibitor | −65.92 | −86.80 | −69.40 |
| fludroxycortide | Glucocorticoid receptor agonist | −69.60 | −58.68 | −67.78 |
| dexamethasone | Glucocorticoid receptor agonist | −63.63 | −86.38 | −67.42 |
| nitrendipine | Calcium channel blocker | −70.41 | −52.10 | −67.36 |
| acyclovir | DNA polymerase inhibitor | −64.90 | −71.25 | −65.96 |
| DMAB-anabaseine | Adrenergic receptor agonist | −68.48 | −51.34 | −65.62 |
| m-chlorophenylbiguanide | Serotonin receptor agonist | −61.73 | −82.67 | −65.22 |
| SB-590885 | RAF inhibitor | −65.27 | −59.71 | −64.34 |
| trimebutine | Opioid receptor agonist | −58.98 | −84.10 | −63.17 |
| teicoplanin | Bacterial cell wall synthesis inhibitor | −64.10 | −56.12 | −62.77 |
| PD-102807 | Acetylcholine receptor antagonist | −61.37 | −61.17 | −61.34 |
| benzatropine | Acetylcholine receptor antagonist | −55.99 | −81.83 | −60.30 |
| GBR-13069 | Dopamine uptake inhibitor | −57.34 | −70.82 | −59.59 |
| vinblastine | Microtubule inhibitor | −55.40 | −76.83 | −58.97 |
| sulindac | Cyclooxygenase inhibitor | −58.98 | −55.76 | −58.44 |
| dexketoprofen | Cyclooxygenase inhibitor | −59.67 | −50.92 | −58.21 |
| hydrocortisone | Glucocorticoid receptor agonist | −50.67 | −91.73 | −57.51 |
| fludrocortisone | Glucocorticoid receptor agonist | −50.42 | −92.94 | −57.51 |
| OMDM-2 | FAAH inhibitor | −57.04 | −58.07 | −57.21 |
| diflorasone | Corticosteroid agonist | −54.44 | −70.40 | −57.10 |
| beclometasone | Glucocorticoid receptor agonist | −52.13 | −72.93 | −55.60 |
| marbofloxacin | Bacterial DNA gyrase inhibitor | −55.04 | −51.70 | −54.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, A.L.; Panganiban, R.; Qiu, W.; Kho, A.T.; Chupp, G.; Meyers, D.A.; Bleecker, E.R.; Weiss, S.T.; Lu, Q.; Tantisira, K.G. Drug Repurposing to Treat Glucocorticoid Resistance in Asthma. J. Pers. Med. 2021, 11, 175. https://doi.org/10.3390/jpm11030175
Wang AL, Panganiban R, Qiu W, Kho AT, Chupp G, Meyers DA, Bleecker ER, Weiss ST, Lu Q, Tantisira KG. Drug Repurposing to Treat Glucocorticoid Resistance in Asthma. Journal of Personalized Medicine. 2021; 11(3):175. https://doi.org/10.3390/jpm11030175
Chicago/Turabian StyleWang, Alberta L., Ronald Panganiban, Weiliang Qiu, Alvin T. Kho, Geoffrey Chupp, Deborah A. Meyers, Eugene R. Bleecker, Scott T. Weiss, Quan Lu, and Kelan G. Tantisira. 2021. "Drug Repurposing to Treat Glucocorticoid Resistance in Asthma" Journal of Personalized Medicine 11, no. 3: 175. https://doi.org/10.3390/jpm11030175
APA StyleWang, A. L., Panganiban, R., Qiu, W., Kho, A. T., Chupp, G., Meyers, D. A., Bleecker, E. R., Weiss, S. T., Lu, Q., & Tantisira, K. G. (2021). Drug Repurposing to Treat Glucocorticoid Resistance in Asthma. Journal of Personalized Medicine, 11(3), 175. https://doi.org/10.3390/jpm11030175