
Targeted Sequencing of Taiwanese Breast Cancer with Risk Stratification by the Concurrent Genes Signature: A Feasibility Study


Abstract
:1. Introduction
2. Materials and Methods
2.1. Overall Aims
2.1.1. Breast Cancer Sample Recruitment
2.1.2. Nucleic Acid Extraction for GE Microarray and NanoString nCounter
2.1.3. Nucleic Acid Extraction for Targeted Sequencing
2.1.4. Extended Concurrent Genes Signature
2.1.5. Actionable Genes for Targeted Sequencing
2.1.6. Library Preparation and NGS Experiments
2.1.7. Variant Annotation and Statistical Analysis
2.1.8. Data Availability
3. Results
3.1. Actionable Genes for Targeted Sequencing
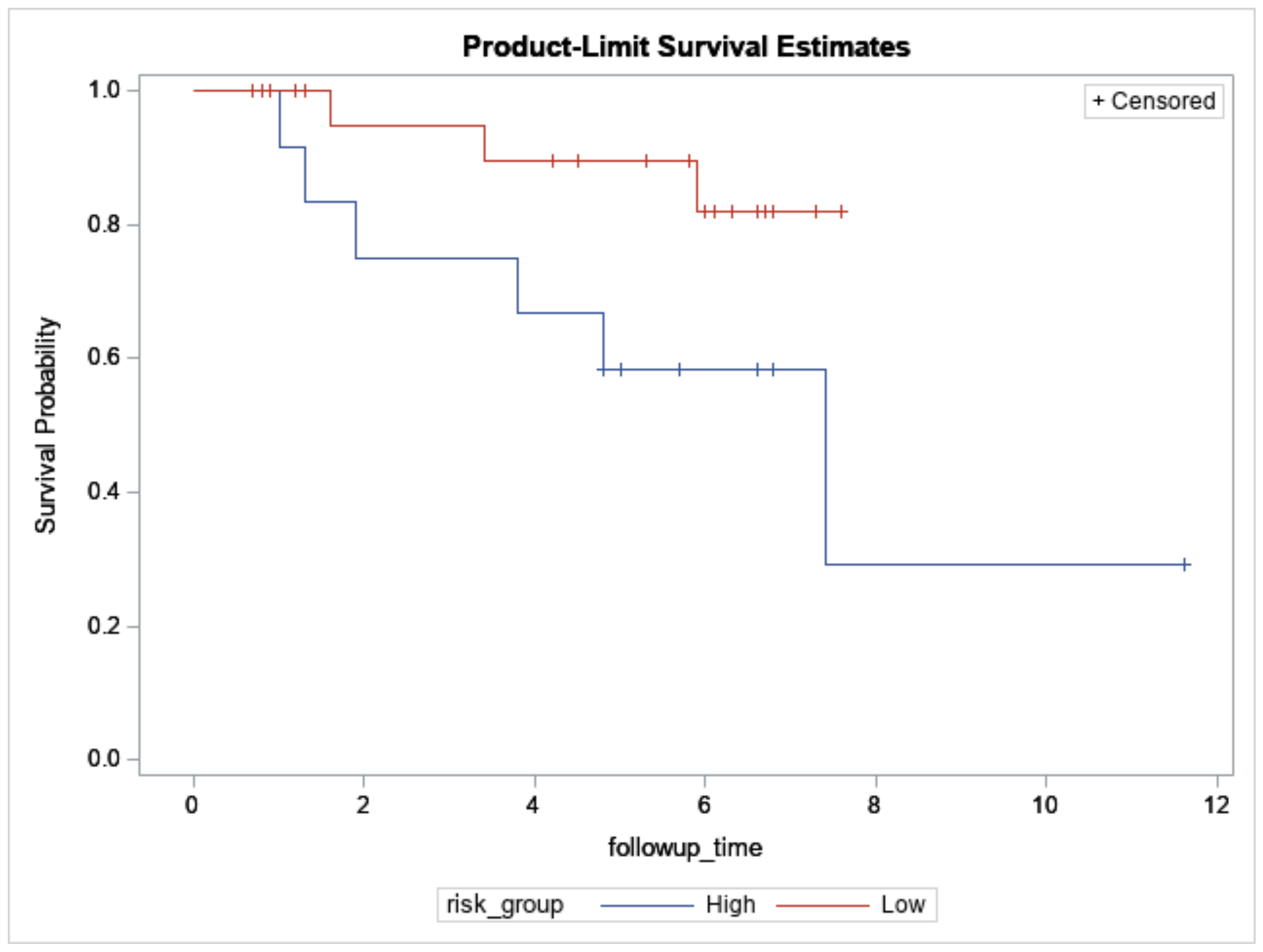
3.2. Breast Cancers Assayed for Targeted Sequencing
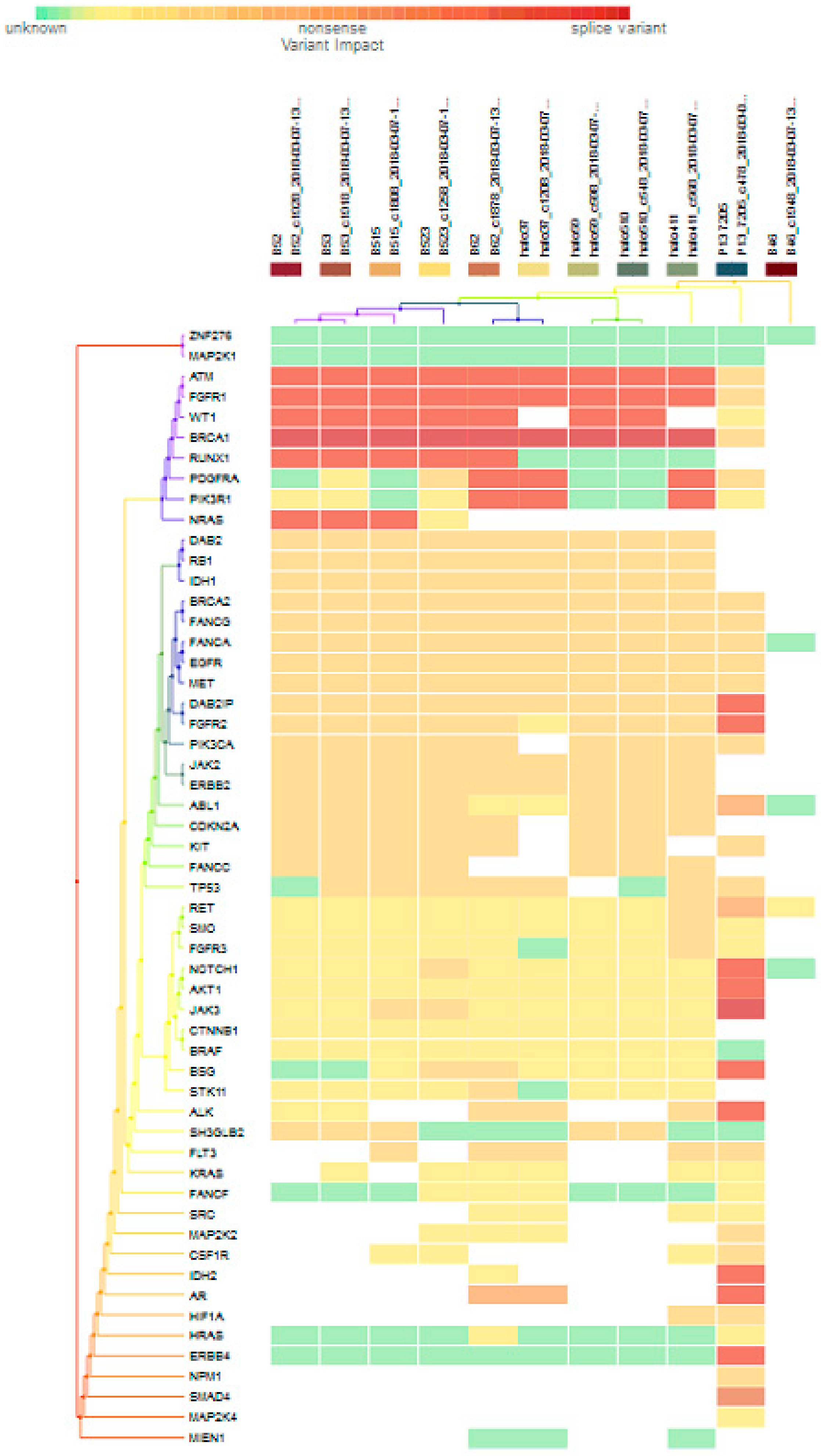
3.3. Significantly Mutated Genes between the High- and Low-Risk Breast Cancers
3.4. Variants-Associated Differentially Expressed Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ministry of Health and Welfare, Taiwan. Statistics of Causes of Death. Available online: www.mohw.gov.tw/cp-16-48057-1.html (accessed on 19 March 2020).
- Yen, A.M.-F.; Tsau, H.-S.; Fann, J.C.-Y.; Chen, S.L.-S.; Chiu, S.Y.-H.; Lee, Y.-C.; Pan, S.-L.; Chiu, H.-M.; Kuo, W.-H.; Chang, K.-J.; et al. Population-Based Breast Cancer Screening with Risk-Based and Universal Mammography Screening Compared with Clinical Breast Examination. JAMA Oncol. 2016, 2, 915–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Arranz, E.E.; Vara, J.Á.F.; Gámez-Pozo, A.; Zamora, P. Gene signatures in breast cancer: Current and future uses. Transl. Oncol. 2012, 5, 398–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slamon, D.; Eiermann, W.; Robert, N.; Giermek, J.; Martin, M.; Jasiowka, M.; Mackey, J.; Chan, A.; Liu, M.-C.; Pinter, T.; et al. Abstract S5-04: Ten year follow-up of BCIRG-006 comparing doxorubicin plus cyclophosphamide followed by docetaxel (AC→T) with doxorubicin plus cyclophosphamide followed by docetaxel and trastuzumab (AC→TH) with docetaxel, carboplatin and trastuzumab (TCH) in HER2+ early breast cancer. Gen. Sess. Abstr. 2016, 76, S5-04. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.; Oh, D.S.; Wessels, L.; Weigelt, B.; Nuyten, D.S.; Nobel, A.B.; Veer, L.J.V.; Perou, C.M. Concordance among Gene-Expression–Based Predictors for Breast Cancer. N. Engl. J. Med. 2006, 355, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.A.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Huang, C.C.; Tu, S.H.; Lien, H.H.; Jeng, J.Y.; Huang, C.S.; Huang, C.J.; Lai, L.C.; Chuang, E.Y. Concurrent gene signatures for han chinese breast cancers. PLoS ONE. 2013, 8, e76421. [Google Scholar] [CrossRef] [Green Version]
- Kao, K.-J.; Chang, K.-M.; Hsu, H.-C.; Huang, A.T. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: Implications for treatment optimization. BMC Cancer 2011, 11, 143. [Google Scholar] [CrossRef] [Green Version]
- Xuan, J.; Yu, Y.; Qing, T.; Guo, L.; Shi, L. Next-generation sequencing in the clinic: Promises and challenges. Cancer Lett. 2013, 340, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Rehm, H.L. Disease-targeted sequencing: A cornerstone in the clinic. Nat. Rev. Genet. 2013, 14, 295–300. [Google Scholar] [CrossRef]
- Huang, C.-S.; Lu, T.-P.; Liu, C.-Y.; Huang, C.-C.; Chiu, J.-H.; Chen, Y.-J.; Tseng, L.-M. Residual risk stratification of Taiwanese breast cancers following curative therapies with the extended concurrent genes signature. Breast Cancer Res. Treat. 2021, 186, 475–485. [Google Scholar] [CrossRef]
- Huang, C.C.; Tu, S.H.; Huang, C.S.; Lien, H.H.; Lai, L.C.; Chuang, E.Y. Abstract 1400: Extended concurrent gene signatures of ER, HER2 and disease-free survival in breast cancers. Cancer Res. 2014, 74 (Suppl. 19), 1400. [Google Scholar]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.C.; Tu, S.H.; Huang, C.S.; Lien, H.H.; Lai, L.C.; Chuang, E.Y. Multiclass prediction with partial least square regression for gene expression data: Applications in breast cancer intrinsic taxonomy. Biomed. Res. Int. 2013, 2013, 248648. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Smigielski, E.M.; Sirotkin, K.; Ward, M.; Sherry, S.T. dbSNP: A database of single nucleotide polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Brusco, L.; Shaw, K.; Horombe, C.; Kopetz, S.; Davies, M.A.; Routbort, M.J.; Piha-Paul, S.A.; Janku, F.; Ueno, N.T.; et al. Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment onto Genomically Matched Clinical Trials. J. Clin. Oncol. 2015, 33, 2753–2762. [Google Scholar] [CrossRef]
- Arnedos, M.; Vicier, C.; Loi, S.; Lefebvre, C.; Michiels, S.; Bonnefoi, H.; Andre, F. Precision medicine for metastatic breast cancer—limitations and solutions. Nat. Rev. Clin. Oncol. 2015, 12, 693–704. [Google Scholar] [CrossRef]
- Relling, M.V.; Evans, W.E. Pharmacogenomics in the clinic. Nat. Cell Biol. 2015, 526, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grada, A.; Weinbrecht, K. Next-Generation Sequencing: Methodology and Application. J. Investig. Dermatol. 2013, 133, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsongalis, G.J.; Peterson, J.D.; De Abreu, F.B.; Tunkey, C.D.; Gallagher, T.L.; Strausbaugh, L.D.; Wells, W.A.; Amos, C.I. Routine use of the Ion Torrent AmpliSeq™ Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin. Chem. Lab. Med. 2014, 52, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Moens, L.N.; Falk-Sörqvist, E.; Ljungström, V.; Mattsson, J.; Sundström, M.; La Fleur, L.; Mathot, L.; Micke, P.; Nilsson, M.; Botling, J. HaloPlex Targeted Resequencing for Mutation Detection in Clinical Formalin-Fixed, Paraffin-Embedded Tumor Samples. J. Mol. Diagn. 2015, 17, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.; Hunter, C.; Bignell, G.; Edkins, S.; Davies, H.; Teague, J.; Stevens, C.; O’Meara, S.; Smith, R.; Parker, A.; et al. Lung cancer: Intragenic ERBB2 kinase mutations in tumours. Nature 2004, 431, 525–526. [Google Scholar] [CrossRef]
- Torkamani, A.; Schork, N.J. Prediction of Cancer Driver Mutations in Protein Kinases. Cancer Res. 2008, 68, 1675–1682. [Google Scholar] [CrossRef] [Green Version]
- Santolla, M.F.; Maggiolini, M. The FGF/FGFR System in Breast Cancer: Oncogenic Features and Therapeutic Perspectives. Cancers 2020, 12, 3029. [Google Scholar] [CrossRef]
- Turner, N.; Lambros, M.B.; Horlings, H.M.; Pearson, A.; Sharpe, R.; Natrajan, R.; Geyer, F.C.; Van Kouwenhove, M.; Kreike, B.; Mackay, A.; et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 2010, 29, 2013–2023. [Google Scholar] [CrossRef] [Green Version]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. New Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Gonçalves, A.; Rugo, H.S.; Lee, K.; Fehrenbacher, L.; Mina, L.A.; Diab, S.; Blum, J.L.; Chakrabarti, J.; Elmeliegy, M.; et al. Talazoparib in Patients with a Germline BRCA -Mutated Advanced Breast Cancer: Detailed Safety Analyses from the Phase III EMBRACA Trial. Oncologist 2020, 25, e439–e450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. SOLAR-1 Study Group. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Sokolova, A.O.; Shirts, B.H.; Konnick, E.Q.; Tsai, G.J.; Goulart, B.H.L.; Montgomery, B.; Pritchard, C.C.; Yu, E.Y.; Cheng, H.H. Complexities of Next-Generation Sequencing in Solid Tumors: Case Studies. J. Natl. Compr. Canc. Netw. 2020, 18, 1150–1155. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Turturro, S.B.; Najor, M.S.; Yung, T.; Portt, L.; Malarkey, C.; Abukhdeir, A.M.; Cobleigh, M.A. Somatic loss of PIK3R1 may sensitize breast cancer to inhibitors of the MAPK pathway. Breast Cancer Res. Treat. 2019, 177, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, L.; Yao, L.; Kuang, X.-Y.; Zuo, W.-J.; Li, S.; Qiao, F.; Liu, Y.-R.; Cao, Z.-G.; Zhou, S.-L.; et al. Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat. Commun. 2018, 9, 1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierrat, B.; Simonen, M.; Cueto, M.; Mestan, J.; Ferrigno, P.K.; Heim, J. SH3GLB, a New Endophilin-Related Protein Family Featuring an SH3 Domain. Genomics 2001, 71, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Serfass, J.M.; Takahashi, Y.; Zhou, Z.; Kawasawa, Y.I.; Liu, Y.; Tsotakos, N.; Young, M.M.; Tang, Z.; Yang, L.; Atkinson, J.M.; et al. Endophilin B2 facilitates endosome maturation in response to growth factor stimulation, autophagy induction, and influenza A virus infection. J. Biol. Chem. 2017, 292, 10097–10111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, J.M.; Guerriero, J.L.; Liu, J.; Ullman, E.; Sheshadri, N.; Chen, J.J.; Zong, W.-X. Elevated Expression of Squamous Cell Carcinoma Antigen (SCCA) Is Associated with Human Breast Carcinoma. PLoS ONE 2011, 6, e19096. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.-Y.; Tai, Y.-L.; Shen, T.-L. Grb7, a Critical Mediator of EGFR/ErbB Signaling, in Cancer Development and as a Potential Therapeutic Target. Cells 2019, 8, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidyanathan, S.; Cato, K.; Tang, L.; Pavey, S.; Haass, N.; Gabrielli, B.G.; Duijf, P.H.G. In vivo overexpression of Emi1 promotes chromosome instability and tumorigenesis. Oncogene 2016, 35, 5446–5455. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Follow-Up Time (Year) | Relapse Status * | Vital Status * | Age | Stage | Predicted Risk Group | HR | HER2 | Grade |
|---|---|---|---|---|---|---|---|---|---|
| #36 | 0.9 | 0 | 1 | 68 | 0 | 1 | 0 | 3 | |
| #38 | 0.7 | 0 | 1 | 55 | 1A | 1 | 0 | 1 | |
| #46 | 4.8 | 1 | 1 | 47 | 3A | High | 1 | 1 | 3 |
| #51 | 1.3 | 0 | 0 | 58 | 4 | High | 0 | 1 | 3 |
| #52 | 6.8 | 0 | 1 | 55 | 3C | Low | 1 | 1 | 3 |
| #53 | 6.7 | 0 | 1 | 34 | 1 | Low | 1 | 1 | 2 |
| #55 | 5.0 | 0 | 1 | 47 | 0 | High | 1 | 0 | 2 |
| #57 | 6.7 | 0 | 1 | 57 | 1 | Low | 1 | 0 | 2 |
| #61 | 0.8 | 0 | 1 | 48 | 3A | Low | 1 | 0 | 3 |
| #62 | 3.9 | 1 | 0 | 56 | 3A | 1 | 1 | 3 | |
| #63 | 0.8 | 0 | 1 | 55 | 2B | 1 | 0 | 3 | |
| #64 | 0.9 | 0 | 1 | 61 | 2A | Low | 1 | 0 | 2 |
| #65 | 0.7 | 1 | 1 | 67 | 3C | Low | 1 | 0 | 2 |
| #511 | 6.1 | 0 | 1 | 38 | 2A | Low | 0 | 1 | 2 |
| #513 | 6.0 | 0 | 1 | 55 | 1 | Low | 0 | 1 | 2 |
| #514 | 5.8 | 0 | 1 | 44 | 0 | Low | 1 | 0 | 3 |
| #515 | 5.7 | 0 | 1 | 47 | 2B | High | 1 | 1 | 3 |
| #517 | 4.2 | 0 | 1 | 52 | 0 | Low | 0 | 1 | 3 |
| #518 | 4.5 | 0 | 1 | 48 | 1 | Low | 1 | 0 | 2 |
| #519 | 1.3 | 0 | 1 | 42 | 2B | Low | 0 | 1 | 3 |
| #520 | 7.4 | 0 | 0 | 74 | 1 | High | 1 | 0 | 3 |
| #521 | 1.9 | 0 | 0 | 44 | 2A | High | 0 | 0 | 3 |
| #522 | 4.8 | 1 | 0 | 39 | 4 | High | 1 | 0 | 2 |
| #523 | 5.3 | 0 | 1 | 53 | 2B | Low | 1 | 1 | 2 |
| #31 | 1.0 | 0 | 1 | 45 | 2B | 1 | 0 | 3 | |
| #32 | 1.0 | 1 | 0 | 46 | 4 | High | 0 | 1 | 3 |
| #33 | 11.6 | 1 | 1 | 50 | 3B | High | 1 | 0 | 3 |
| #35 | 0.9 | 0 | 1 | 70 | 2A | 1 | 0 | 2 | |
| #37 | 0.8 | 0 | 1 | 51 | 1A | 1 | 1 | 1 | |
| #39 | 0.8 | 0 | 1 | 44 | 1A | 1 | 0 | 2 | |
| #41 | 5.8 | 0 | 1 | 50 | 1 | Low | 1 | 0 | 3 |
| #42 | 1.6 | 1 | 0 | 50 | 4 | Low | 0 | 1 | 3 |
| #43 | 7.6 | 0 | 1 | 33 | 2B | Low | 1 | 0 | 1 |
| #44 | 7.3 | 0 | 1 | 54 | 3A | Low | 0 | 0 | 3 |
| #45 | 5.9 | 1 | 0 | 69 | 2B | Low | 0 | 0 | 2 |
| #47 | 3.4 | 1 | 0 | 42 | 2A | Low | 1 | 0 | 3 |
| #48 | 3.8 | 0 | 0 | 57 | 2B | High | 0 | 1 | 3 |
| #49 | 1.2 | 1 | 1 | 57 | 999 | Low | 1 | 0 | 2 |
| #54 | 6.8 | 0 | 1 | 43 | 2A | High | 0 | 0 | 3 |
| #56 | 6.6 | 0 | 1 | 46 | 2B | High | 0 | 1 | 3 |
| #58 | 6.8 | 0 | 1 | 45 | 2A | Low | 0 | 0 | 3 |
| #59 | 6.6 | 0 | 1 | 46 | 1 | Low | 1 | 1 | 2 |
| #310 | 0.8 | 0 | 1 | 59 | 1A | 0 | 0 | 3 | |
| #410 | 0.8 | 0 | 1 | 59 | 1A | Low | 0 | 0 | 3 |
| #411 | 0.8 | 0 | 1 | 50 | 3C | Low | 1 | 1 | 3 |
| #412 | 0.9 | 0 | 1 | 64 | 1A | 0 | 1 | 3 | |
| #510 | 6.3 | 0 | 1 | 69 | 2A | Low | 1 | 1 | 2 |
| #512 | 6.1 | 1 | 1 | 61 | 2A | Low | 1 | 0 | 2 |
| #13 | 1.0 | 0 | 1 | 47 | 1A | 1 | 0 | 2 | |
| #16 | 0.8 | 0 | 1 | 46 | 0 | 1 | 0 | 2 | |
| #19 | 0.3 | 0 | 1 | 56 | 1A | 1 | 0 | 1 | |
| #18 | 0.9 | 0 | 1 | 45 | 2A | 1 | 0 | 2 | |
| #14 | 5.3 | 0 | 1 | 39 | 2A | 0 | 0 | 3 | |
| #12 | 0.1 | 0 | 1 | 62 | 3C | 1 | 1 | 2 | |
| #110 | 6.0 | 0 | 1 | 63 | 0 | 1 | 0 | 1 | |
| #11 | 1.0 | 0 | 1 | 37 | 1A | 1 | 0 | 1 | |
| #21 | 0.8 | 0 | 1 | 58 | 3A | 1 | 0 | 1 | |
| #22 | 0.9 | 0 | 1 | 46 | 1A | 0 | 0 | 3 | |
| #23 | 0.8 | 0 | 1 | 60 | 2A | 1 | 0 | 1 | |
| #24 | 0.9 | 0 | 1 | 88 | 1A | 1 | 0 | 2 | |
| #25 | 0.9 | 0 | 1 | 50 | 2B | 0 | 0 | 3 |
| Gene | refSNP ID | Type | Function Class | Cosmic Amino Acid Syntax | Impacted Patients |
|---|---|---|---|---|---|
| ERBB2 | rs28933370 | SNP | MISSENSE | p.N857S | 46 |
| PIK3CA | rs121913279 | SNP | MISSENSE | p.H1047L,p.H1047R,p.H1047P | 8 |
| BRCA2 | Deletion | p.I605fs*9 | 6 | ||
| TP53 | rs11540652 | SNP | MISSENSE | p.R248Q,p.R248L,p.R248P,p.R155Q,p.R155P,p.R155L | 3 |
| CTNNB1 | SNP | NONSENSE | 1 | ||
| FGFR3 | rs121913112 | SNP | MISSENSE | 1 | |
| CSF1R | SNP | MISSENSE | 1 | ||
| JAK2 | rs77375493 | SNP | MISSENSE | p.V617F,p.V617I,p.V617_C618 > FR | 1 |
| HRAS | rs104894228 | SNP | MISSENSE | p.G13R,p.G13S,p.G13C | 1 |
| TP53 | SNP | NONSENSE | p.R306* | 1 | |
| TP53 | rs28934578 | SNP | MISSENSE | p.R175H,p.R175L,p.R43H,p.R82H,p.R82L,p.R175P,p.R43L | 1 |
| RUNX1 | SNP | MISSENSE | p.H85N | 1 |
| Gene | Mutation Type | refSNP ID | ACMG Category | Function Class | p-Value (χ2-Test) |
|---|---|---|---|---|---|
| PIK3CA | SNP | Category II | MISSENSE | 0.03 | |
| PIK3CA | SNP | rs121913279 | Category II | MISSENSE | 0.02 |
| PDGFRA | SNP | rs35597368 | Category II | MISSENSE | 0.01 |
| CSF1R | SNP | Category III | 0.02 | ||
| EGFR | SNP | Category III | SILENT | 0.02 | |
| MET | SNP | rs41736 | Category III | SILENT | 0.05 |
| FGFR1 | SNP | Category III | 0.01 | ||
| SH3GLB2 | SNP | Category III | 0.02 | ||
| SH3GLB2 | SNP | Category II | MISSENSE | 0.02 | |
| ATM | Deletion | Category I | 0.04 | ||
| BRCA2 | SNP | rs56403624 | Category II | MISSENSE | 0.02 |
| BRCA2 | SNP | rs169547 | Category II | MISSENSE | 0.04 |
| FANCA | SNP | Category III | SILENT | 0.01 | |
| FANCA | SNP | Category III | 0.04 | ||
| ERBB2 | SNP | Category III | SILENT | 0.02 | |
| ERBB2 | SNP | Category II | MISSENSE | 0.02 | |
| BRCA1 | SNP | rs55946644 | Category III | 0.01 | |
| BSG | SNP | Category III | 0.02 | ||
| BSG | SNP | Category III | SILENT | 0.04 | |
| BSG | SNP | Category III | SILENT | 0.01 | |
| MAP2K2 | SNP | rs10250 | Category III | SILENT | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.-S.; Liu, C.-Y.; Lu, T.-P.; Huang, C.-J.; Chiu, J.-H.; Tseng, L.-M.; Huang, C.-C. Targeted Sequencing of Taiwanese Breast Cancer with Risk Stratification by the Concurrent Genes Signature: A Feasibility Study. J. Pers. Med. 2021, 11, 613. https://doi.org/10.3390/jpm11070613
Huang C-S, Liu C-Y, Lu T-P, Huang C-J, Chiu J-H, Tseng L-M, Huang C-C. Targeted Sequencing of Taiwanese Breast Cancer with Risk Stratification by the Concurrent Genes Signature: A Feasibility Study. Journal of Personalized Medicine. 2021; 11(7):613. https://doi.org/10.3390/jpm11070613
Chicago/Turabian StyleHuang, Ching-Shui, Chih-Yi Liu, Tzu-Pin Lu, Chi-Jung Huang, Jen-Hwey Chiu, Ling-Ming Tseng, and Chi-Cheng Huang. 2021. "Targeted Sequencing of Taiwanese Breast Cancer with Risk Stratification by the Concurrent Genes Signature: A Feasibility Study" Journal of Personalized Medicine 11, no. 7: 613. https://doi.org/10.3390/jpm11070613
APA StyleHuang, C. -S., Liu, C. -Y., Lu, T. -P., Huang, C. -J., Chiu, J. -H., Tseng, L. -M., & Huang, C. -C. (2021). Targeted Sequencing of Taiwanese Breast Cancer with Risk Stratification by the Concurrent Genes Signature: A Feasibility Study. Journal of Personalized Medicine, 11(7), 613. https://doi.org/10.3390/jpm11070613