
Ultrarare Loss-of-Function Mutations in the Genes Encoding the Ionotropic Glutamate Receptors of Kainate Subtypes Associated with Schizophrenia Disrupt the Interaction with PSD95

 ,
,
Abstract
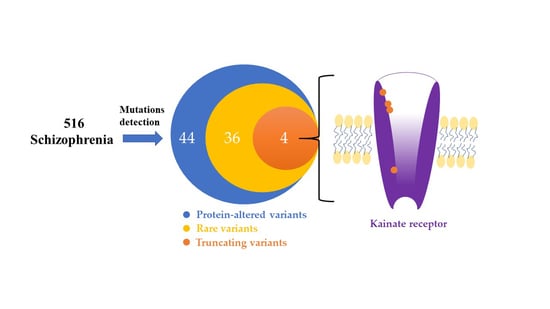
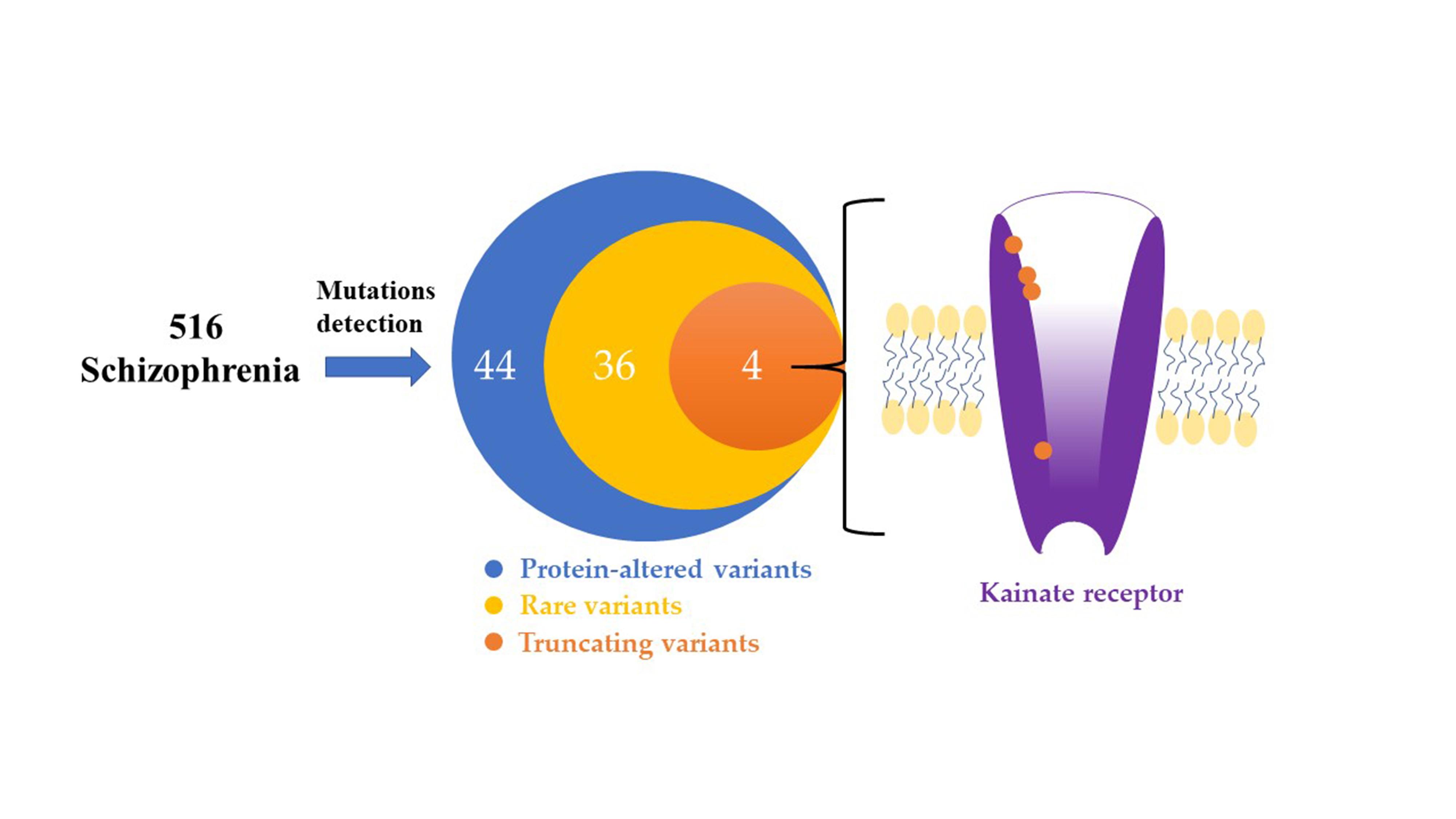
:
1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Ion Semiconductor Sequencing, PCR Reaction, and Fluorescence-Based Cycle Sequencing
2.3. In Silico Analysis of Amino Acid Substitutions
2.4. RNA Preparation and Semi-Quantitative RT-PCR
2.5. Gene Constructs and Transient Transfection of Cell Lines
2.6. Immunoblot Assay
2.7. Immunocytochemistry
2.8. BRET Assay
3. Results
3.1. Identification of the Rare and Protein-Altering Variants of the GRIK Gene Family in Patients with Schizophrenia
3.2. GRIK Gene Family mRNA Expressions in Peripheral Blood Cells, Lymphoblastoid Cells, and Human Brain
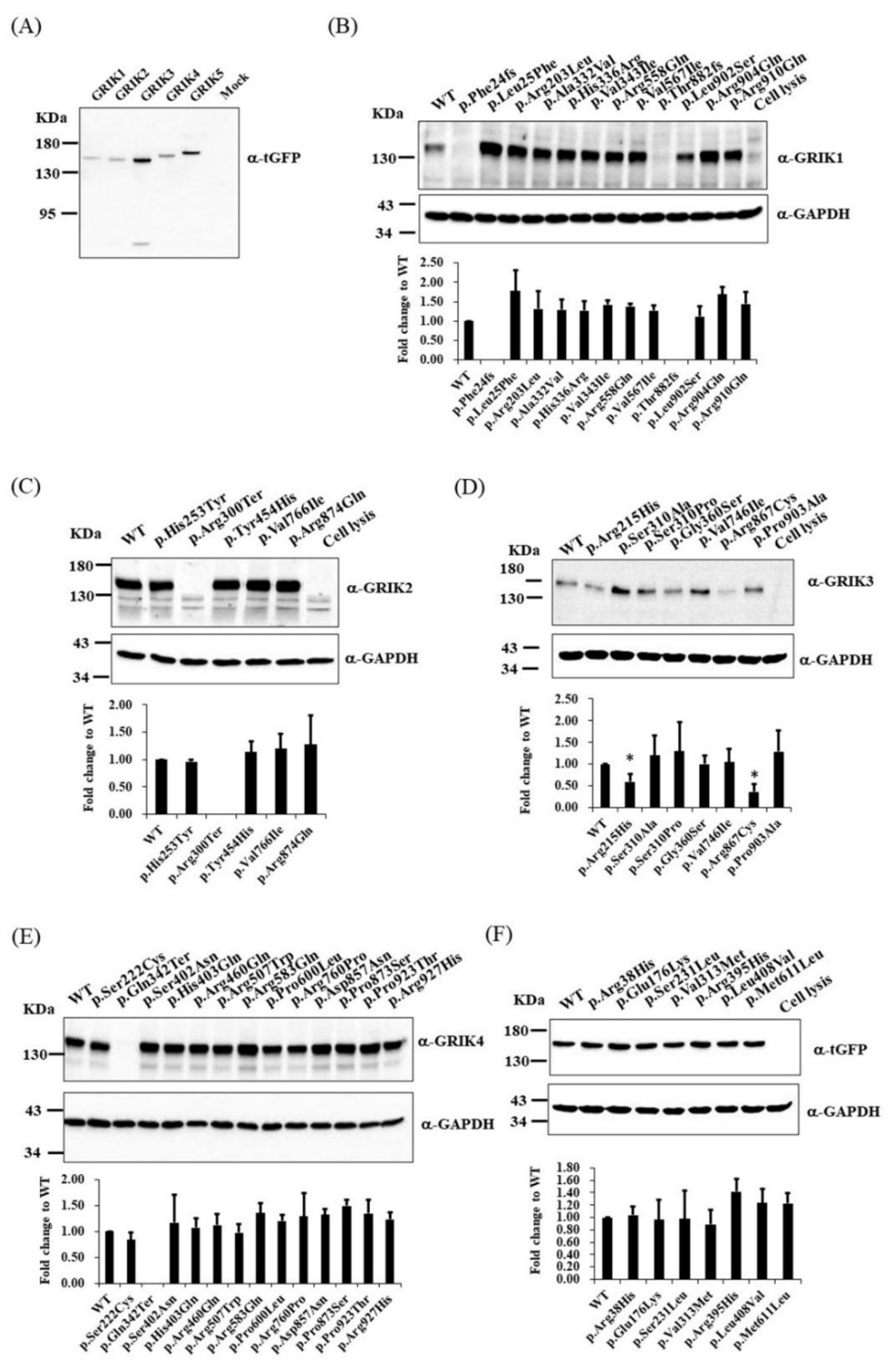
3.3. Immunoblotting and Immunocytochemistry Analysis of GRIK Gene Family Mutants in Cultured Cells
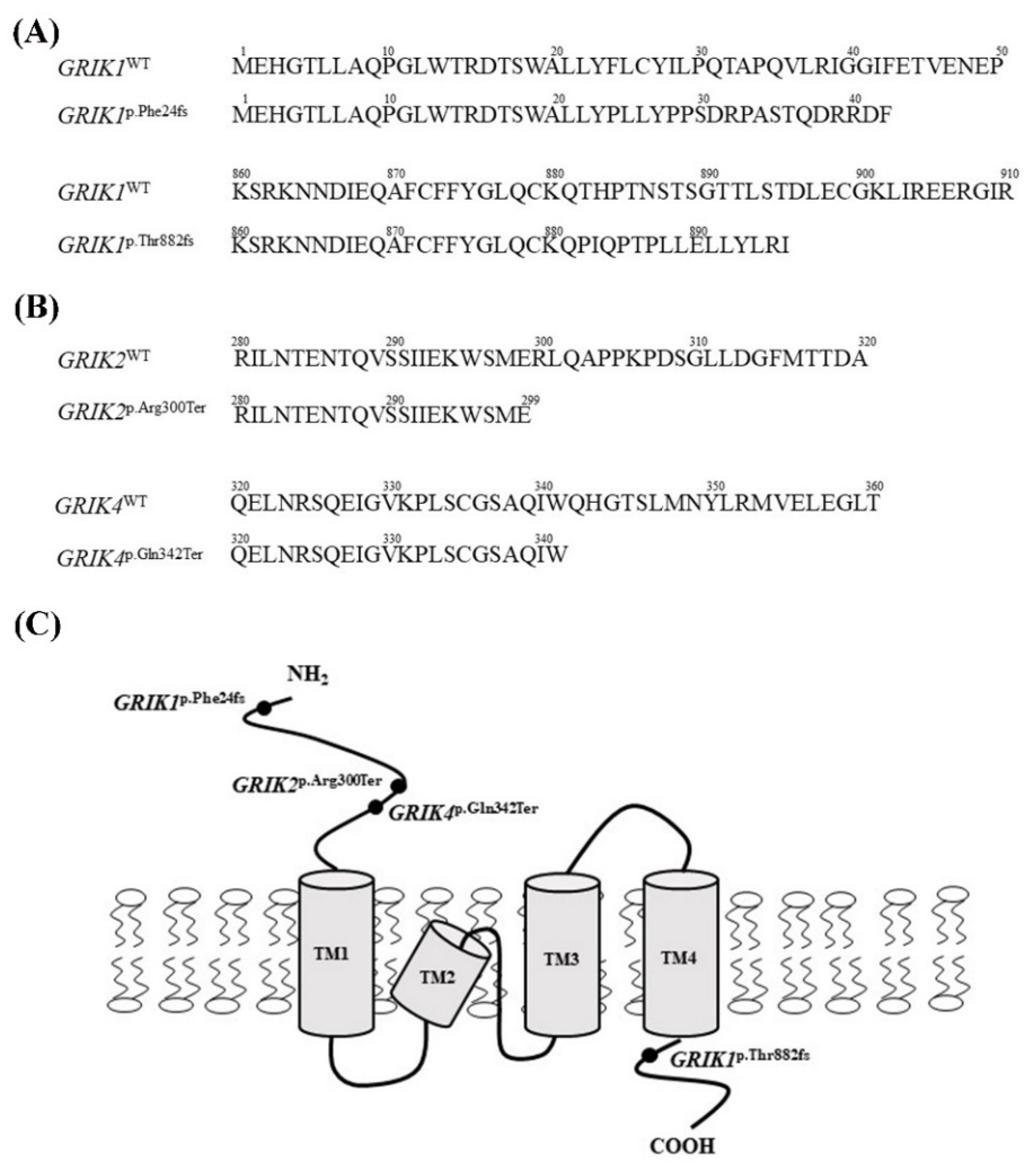
3.4. Protein-Truncating Mutations in the GRIK Gene Family Disrupt the Interaction with the Post-Synaptic Density Protein 95 (PSD95) Protein
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Freedman, R. Schizophrenia. N. Engl. J. Med. 2003, 349, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, D.R. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 1987, 44, 660–669. [Google Scholar] [CrossRef] [PubMed]
- McGlashan, T.H.; Hoffman, R.E. Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch. Gen. Psychiatry 2000, 57, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Stephan, K.E.; Baldeweg, T.; Friston, K.J. Synaptic plasticity and dysconnection in schizophrenia. Biol. Psychiatry 2006, 59, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef]
- van Berlekom, A.B.; Muflihah, C.H.; Snijders, G.; MacGillavry, H.D.; Middeldorp, J.; Hol, E.M.; Kahn, R.S.; de Witte, L.D. Synapse pathology in schizophrenia: A meta-analysis of postsynaptic elements in postmortem brain studies. Schizophr. Bull. 2020, 46, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Belsham, B. Glutamate and its role in psychiatric illness. Hum. Psychopharmacol. 2001, 16, 139–146. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Bortolotto, Z.A.; Clarke, V.R.; Delany, C.M.; Parry, M.C.; Smolders, I.; Vignes, M.; Ho, K.H.; Miu, P.; Brinton, B.T.; Fantaske, R.; et al. Kainate receptors are involved in synaptic plasticity. Nature 1999, 402, 297–301. [Google Scholar] [CrossRef]
- Hollmann, M.; Heinemann, S. Cloned glutamate receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Olsen, R.W.; Peters, J.; Spedding, M. A nomenclature for ligand-gated ion channels. Neuropharmacology 2009, 56, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamboj, R.K.; Schoepp, D.D.; Nutt, S.; Shekter, L.; Korczak, B.; True, R.A.; Rampersad, V.; Zimmerman, D.M.; Wosnick, M.A. Molecular cloning, expression, and pharmacological characterization of humEAA1, a human kainate receptor subunit. J. Neurochem. 1994, 62, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lowry, E.R.; Kruyer, A.; Norris, E.H.; Cederroth, C.R.; Strickland, S. The GluK4 kainate receptor subunit regulates memory, mood, and excitotoxic neurodegeneration. Neuroscience 2013, 235, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, V.; Pecoraro, V.; Aller, M.I.; Roman, C.; Paternain, A.V.; Lerma, J. Increased Grik4 gene dosage causes imbalanced circuit output and human disease-related behaviors. Cell Rep. 2018, 23, 3827–3838. [Google Scholar] [CrossRef]
- Mueller, H.T.; Haroutunian, V.; Davis, K.L.; Meador-Woodruff, J.H. Expression of the ionotropic glutamate receptor subunits and NMDA receptor-associated intracellular proteins in the substantia nigra in schizophrenia. Mol. Brain Res. 2004, 121, 60–69. [Google Scholar] [CrossRef]
- Scarr, E.; Beneyto, M.; Meador-Woodruff, J.H.; Dean, B. Cortical glutamatergic markers in schizophrenia. Neuropsychopharmacology 2005, 30, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Beneyto, M.; Kristiansen, L.V.; Oni-Orisan, A.; McCullumsmith, R.E.; Meador-Woodruff, J.H. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 2007, 32, 1888–1902. [Google Scholar] [CrossRef]
- Meador-Woodruff, J.H.; Davis, K.L.; Haroutunian, V. Abnormal kainate receptor expression in prefrontal cortex in schizophrenia. Neuropsychopharmacology 2001, 24, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Porter, R.H.; Eastwood, S.L.; Harrison, P.J. Distribution of kainate receptor subunit mrnas in human hippocampus, neocortex and cerebellum, and bilateral reduction of hippocampal GluR6 and KA2 transcripts in schizophrenia. Brain Res. 1997, 751, 217–231. [Google Scholar] [CrossRef]
- Sokolov, B.P. Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mrnas is decreased in frontal cortex of “neuroleptic-free” schizophrenics: Evidence on reversible up-regulation by typical neuroleptics. J. Neurochem. 1998, 71, 2454–2464. [Google Scholar] [CrossRef]
- Ibrahim, H.M.; Hogg, A.J., Jr.; Healy, D.J.; Haroutunian, V.; Davis, K.L.; Meador-Woodruff, J.H. Ionotropic glutamate receptor binding and subunit mRNA expression in thalamic nuclei in schizophrenia. Am. J. Psychiatry 2000, 157, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Garey, L.J.; Von Bussmann, K.A.; Hirsch, S.R. Decreased numerical density of kainate receptor-positive neurons in the orbitofrontal cortex of chronic schizophrenics. Exp. Brain Res. 2006, 173, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Todtenkopf, M.S.; Kostoulakos, P. GluR5,6,7 subunit immunoreactivity on apical pyramidal cell dendrites in hippocampus of schizophrenics and manic depressives. Hippocampus 2001, 11, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Joo, A.; Fujii, Y.; Tani, A.; Makino, C.; Hirata, N.; Kikuta, R.; Ninomiya, H.; Tashiro, N.; Fukumaki, Y. Association study of polymorphisms in the glur5 kainate receptor gene (GRIK1) with schizophrenia. Psychiatr. Genet. 2001, 11, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Zai, C.C.; Souza, R.P.; Lieberman, J.A.; Meltzer, H.Y.; Kennedy, J.L. Association study of GRIK1 gene polymorphisms in schizophrenia: Case-control and family-based studies. Hum. Psychopharmacol. 2012, 27, 345–351. [Google Scholar] [CrossRef]
- Shibata, H.; Aramaki, T.; Sakai, M.; Ninomiya, H.; Tashiro, N.; Iwata, N.; Ozaki, N.; Fukumaki, Y. Association study of polymorphisms in the glur7, ka1 and ka2 kainate receptor genes (GRIK3, GRIK4, GRIK5) with schizophrenia. Psychiatry Res. 2006, 141, 39–51. [Google Scholar] [CrossRef]
- Li, Z.; He, Z.; Tang, W.; Tang, R.; Huang, K.; Xu, Z.; Xu, Y.; Li, L.; Li, X.; Feng, G.; et al. No genetic association between polymorphisms in the kainate-type glutamate receptor gene, GRIK4, and schizophrenia in the chinese population. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 876–880. [Google Scholar] [CrossRef]
- Ren, D.; Xu, F.; Bi, Y.; Niu, W.; Zhang, R.; Hu, J.; Guo, Z.; Cao, Y.; Huang, X.; Wu, X.; et al. No association of GRIK4 polymorphisms with schizophrenia in the chinese han population. Psychiatr. Genet. 2017, 27, 159–160. [Google Scholar] [CrossRef]
- Shibata, H.; Shibata, A.; Ninomiya, H.; Tashiro, N.; Fukumaki, Y. Association study of polymorphisms in the glur6 kainate receptor gene (GRIK2) with schizophrenia. Psychiatry Res. 2002, 113, 59–67. [Google Scholar] [CrossRef]
- Lai, I.C.; Liou, Y.J.; Chen, J.Y.; Wang, Y.C. No association between the ionotropic glutamate receptor kainate 3 gene ser310ala polymorphism and schizophrenia. Neuropsychobiology 2005, 51, 211–213. [Google Scholar] [CrossRef]
- Begni, S.; Popoli, M.; Moraschi, S.; Bignotti, S.; Tura, G.B.; Gennarelli, M. Association between the ionotropic glutamate receptor kainate 3 (GRIK3) ser310ala polymorphism and schizophrenia. Mol. Psychiatry 2002, 7, 416–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djurovic, S.; Kahler, A.K.; Kulle, B.; Jonsson, E.G.; Agartz, I.; Le Hellard, S.; Hall, H.; Jakobsen, K.D.; Hansen, T.; Melle, I.; et al. A possible association between schizophrenia and GRIK3 polymorphisms in a multicenter sample of scandinavian origin (SCOPE). Schizophr. Res. 2009, 107, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Pickard, B.S.; Malloy, M.P.; Christoforou, A.; Thomson, P.A.; Evans, K.L.; Morris, S.W.; Hampson, M.; Porteous, D.J.; Blackwood, D.H.; Muir, W.J. Cytogenetic and genetic evidence supports a role for the kainate-type glutamate receptor gene, GRIK4, in schizophrenia and bipolar disorder. Mol. Psychiatry 2006, 11, 847–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, T.A.; Lazzeroni, L.C.; Calkins, M.E.; Freedman, R.; Green, M.F.; Gur, R.E.; Gur, R.C.; Light, G.A.; Nuechterlein, K.H.; Olincy, A.; et al. Genetic assessment of additional endophenotypes from the consortium on the genetics of schizophrenia family study. Schizophr. Res. 2016, 170, 30–40. [Google Scholar] [CrossRef] [Green Version]
- McClellan, J.; King, M.C. Genomic analysis of mental illness: A changing landscape. JAMA 2010, 303, 2523–2524. [Google Scholar] [CrossRef]
- Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar]
- Kirov, G.; Pocklington, A.J.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo cnv analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2012, 17, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, J.; Champagne, N.; Lafrenière, R.G.; Xiong, L.; Spiegelman, D.; Brustein, E.; Lapointe, M.; Peng, H.; Côté, M.; Noreau, A.; et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 7863–7868. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Fromer, M.; Stahl, E.A.; Ruderfer, D.M.; Chambert, K.; Landen, M.; Moran, J.L.; Purcell, S.M.; Sklar, P.; Sullivan, P.F.; et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 2016, 19, 1433–1441. [Google Scholar] [CrossRef]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014, 506, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.M.; Wang, Y.C.; Wu, C.L.; Hsu, S.H.; Tsai, H.Y.; Cheng, M.C. Multiple rare risk coding variants in postsynaptic density-related genes associated with schizophrenia susceptibility. Front. Genet. 2020, 11, 524258. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.M.; Chen, S.J.; Hsu, S.H.; Cheng, M.C. Functional analyses and effect of DNA methylation on the EGR1 gene in patients with schizophrenia. Psychiatry Res. 2019, 275, 276–282. [Google Scholar] [CrossRef]
- Khajavi, M.; Inoue, K.; Lupski, J.R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 2006, 14, 1074–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Kulozik, A.E. A perfect message: RNA surveillance and nonsense-mediated decay. Cell 1999, 96, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.; Wu, H.; Garner, C.C.; Marshall, J. Molecular mechanisms regulating the differential association of kainate receptor subunits with SAP90/PSD-95 and SAP97. J. Biol. Chem. 2001, 276, 16092–16099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, E.P.; Mehta, S.; Blair, L.A.; Wells, D.G.; Shang, J.; Fukushima, T.; Fallon, J.R.; Garner, C.C.; Marshall, J. SAP90 binds and clusters kainate receptors causing incomplete desensitization. Neuron 1998, 21, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, E.; Kamiya, H. PSD-95 regulates synaptic kainate receptors at mouse hippocampal mossy fiber-CA3 synapses. Neurosci. Res. 2016, 107, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Kimura, H.; Wang, C.; Ishizuka, K.; Kushima, I.; Arioka, Y.; Yoshimi, A.; Nakamura, Y.; Shiino, T.; Oya-Ito, T.; et al. Resequencing and association analysis of six PSD-95-related genes as possible susceptibility genes for schizophrenia and autism spectrum disorders. Sci. Rep. 2016, 6, 27491. [Google Scholar] [CrossRef]
- Cheng, M.C.; Lu, C.L.; Luu, S.U.; Tsai, H.M.; Hsu, S.H.; Chen, T.T.; Chen, C.H. Genetic and functional analysis of the DLG4 gene encoding the post-synaptic density protein 95 in schizophrenia. PLoS ONE 2010, 5, e15107. [Google Scholar] [CrossRef]
- Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; Witte, J.S.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984–994. [Google Scholar]
- Waltereit, R.; Banaschewski, T.; Meyer-Lindenberg, A.; Poustka, L. Interaction of neurodevelopmental pathways and synaptic plasticity in mental retardation, autism spectrum disorder and schizophrenia: Implications for psychiatry. World J. Biol. Psychiatry 2014, 15, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Rees, E.; Creeth, H.D.J.; Hwu, H.G.; Chen, W.J.; Tsuang, M.; Glatt, S.J.; Rey, R.; Kirov, G.; Walters, J.T.R.; Holmans, P.; et al. Schizophrenia, autism spectrum disorders and developmental disorders share specific disruptive coding mutations. Nat. Commun. 2021, 12, 5353. [Google Scholar] [CrossRef] [PubMed]
- Cordoba, M.; Rodriguez, S.; Moron, D.G.; Medina, N.; Kauffman, M.A. Expanding the spectrum of Grik2 mutations: Intellectual disability, behavioural disorder, epilepsy and dystonia. Clin. Genet. 2015, 87, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Guzman, Y.F.; Ramsey, K.; Stolz, J.R.; Craig, D.W.; Huentelman, M.J.; Narayanan, V.; Swanson, G.T. A gain-of-function mutation in the GRIK2 gene causes neurodevelopmental deficits. Neurol Genet. 2017, 3, e129. [Google Scholar] [CrossRef] [Green Version]
- Stolz, J.R.; Foote, K.M.; Veenstra-Knol, H.E.; Pfundt, R.; Ten Broeke, S.W.; de Leeuw, N.; Roht, L.; Pajusalu, S.; Part, R.; Rebane, I.; et al. Clustered mutations in the GRIK2 kainate receptor subunit gene underlie diverse neurodevelopmental disorders. Am. J. Hum. Genet. 2021, 108, 2206. [Google Scholar] [CrossRef]
- Griswold, A.J.; Ma, D.; Cukier, H.N.; Nations, L.D.; Schmidt, M.A.; Chung, R.H.; Jaworski, J.M.; Salyakina, D.; Konidari, I.; Whitehead, P.L.; et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet. 2012, 21, 3513–3523. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.P.; Magalhaes, T.; Conroy, J.M.; Regan, R.; Shah, N.; Anney, R.; Shields, D.C.; Abrahams, B.S.; Almeida, J.; Bacchelli, E.; et al. A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Hum. Genet. 2012, 131, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Jamain, S.; Betancur, C.; Quach, H.; Philippe, A.; Fellous, M.; Giros, B.; Gillberg, C.; Leboyer, M.; Bourgeron, T. Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry 2002, 7, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Takenouchi, T.; Hashida, N.; Torii, C.; Kosaki, R.; Takahashi, T.; Kosaki, K. 1p34.3 deletion involving GRIK3: Further clinical implication of grik family glutamate receptors in the pathogenesis of developmental delay. Am. J. Med. Genet. A 2014, 164, 456–460. [Google Scholar] [CrossRef]
- Koromina, M.; Flitton, M.; Blockley, A.; Mellor, I.R.; Knight, H.M. Damaging coding variants within kainate receptor channel genes are enriched in individuals with schizophrenia, autism and intellectual disabilities. Sci. Rep. 2019, 9, 19215. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
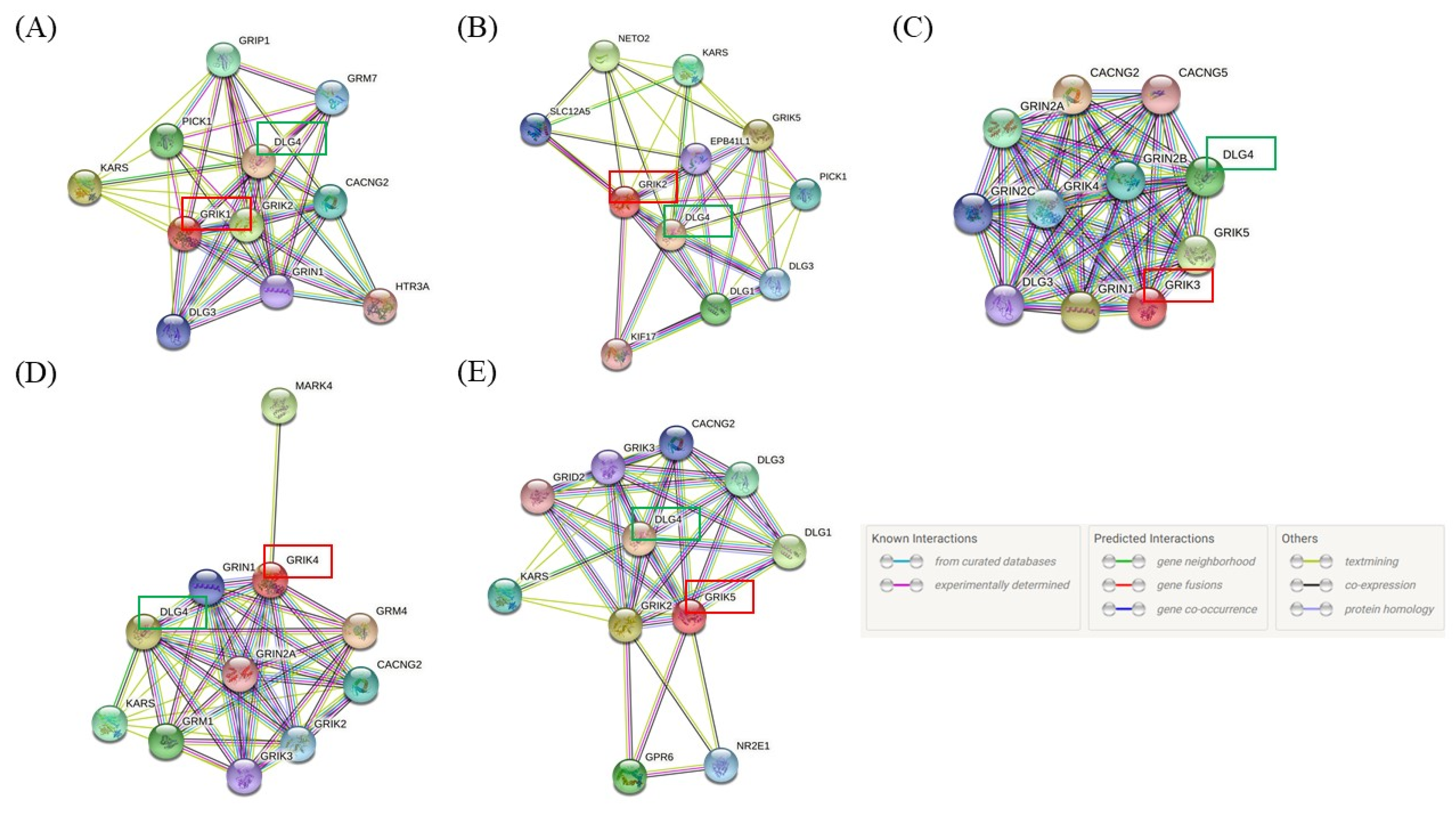{kind=link}
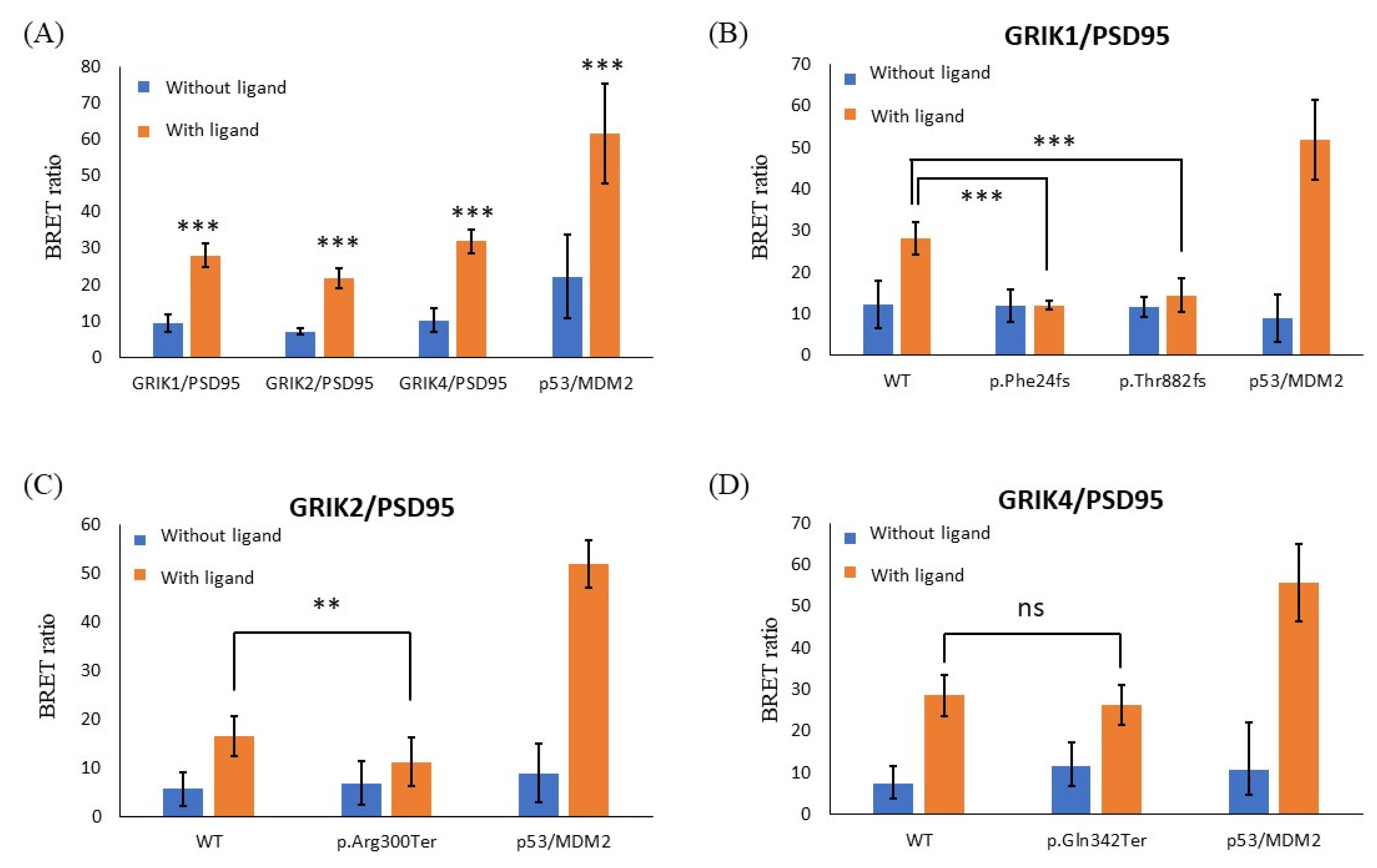{kind=link}
| Genomic Position | Variant (Amino Acid Change) | Patient Number | dbSNP ID | MAF | In Silico Analysis for Amino Acid Substitution | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| gnomAD (Non-Neuro) | 1000G | Taiwan Biobank | Polyphen-2 | SIFT | Pmut | PROVEAN | CADD Raw/PHRED | ||||
| GRIK1 | |||||||||||
| chr21:31311747 | c.70_71delTT (p.Phe24fs) | 1 | N/R | N/R | N/R | N/R | N/A | N/A | N/A | N/A | N/A |
| chr21:31311746 | c.73C > T (p.Leu25Phe) | 2 | rs200262014 | 0.0002 | 0.0008 | 0.0011 | Benign | Tolerated | Neutral | Neutral | 2.15/21.1 |
| chr21:31045421 | c.608G > T (p.Arg203Leu) | 1 | N/R | N/R | N/R | N/R | Probably damaging | Damaging | Disease | Deleterious | 4.27/30 |
| chr21:31015249 | c.995C > T (p.Ala332Val) | 1 | rs143252117 | 0.0001 | 0.0004 | 0.0007 | Benign | Damaging | Disease | Deleterious | 3.56/25.2 |
| chr21:31015237 | c.1007A > G (p.His336Arg) | 1 | rs756781607 | N/R | N/R | N/R | Benign | Tolerated | Neutral | Neutral | 2.51/22.6 |
| chr21:31015217 | c.1027G > A (p.Val343Ile) | 1 | rs1459657790 | <0.0001 | N/R | N/R | Benign | Tolerated | Neutral | Neutral | 2.33/22.1 |
| chr21:30959806 | c.1673G > A (p.Arg558Gln) | 1 | rs776162409 | <0.0001 | N/R | N/R | Probably damaging | Damaging | Disease | Deleterious | 3.90/26.9 |
| chr21:30959780 | c.1699G > A (p.Val567Ile) | 1 | rs536890189 | 0.0001 | 0.0006 | N/R | Probably damaging | Damaging | Disease | Neutral | 3.63/25.5 |
| chr21:30925988 | c.2644delA (p.Thr882fs) | 1 | rs867634999 | <0.0001 | N/R | N/R | N/A | N/A | N/A | N/A | N/A |
| chr21:30925928 | c.2705T > C (p.Leu902Ser) | 41 | rs363504 | 0.0694 | 0.1098 | 0.0639 | Benign | Tolerated | Neutral | Neutral | 2.00/19.76 |
| chr21:30925922 | c.2711G > A (p.Arg904Gln) | 1 | rs769191901 | <0.0001 | N/R | N/R | Benign | Damaging | Neutral | Neutral | 2.62/22.8 |
| chr21:30925904 | c.2729G > A (p.Arg910Gln) | 3 | rs372106416 | 0.0002 | 0.0002 | 0.0023 | Benign | Tolerated | Neutral | Neutral | 2.55/22.6 |
| GRIK2 | |||||||||||
| chr6:102130461 | c.757C > T (p.His253Tyr) | 4 | rs770586258 | <0.0001 | N/R | 0.0007 | Probably damaging | Damaging | Disease | Deleterious | 4.09/28.5 |
| chr6:102134175 | c.898C > T (p.Arg300Ter) | 1 | rs1351417917 | <0.0001 | N/R | N/R | N/A | N/A | N/A | N/A | N/A |
| chr6:102307204 | c.1360T > C (p.Tyr454His) | 8 | rs186727716 | <0.0001 | 0.0006 | 0.0020 | Benign | Tolerated | Neutral | Neutral | 2.29/22 |
| chr6:102483426 | c.2296G > A (p.Val766Ile) | 7 | rs3213608 | 0.0034 | 0.00639 | 0.0026 | Benign | Tolerated | Neutral | Neutral | 2.23/21.6 |
| chr6:102516280 | c.2621G > A (p.Arg874Gln) | 2 | rs267600750 | <0.0001 | N/R | 0.0003 | Possibly damaging | Damaging | Neutral | Neutral | 3.23/24.1 |
| GRIK3 | |||||||||||
| chr1:37337877 | c.644G > A (p.Arg215His) | 1 | rs755366301 | <0.0001 | N/R | N/R | Probably damaging | Damaging | Neutral | Deleterious | 3.90/26.9 |
| chr1:37325477 | c.928T > G (p.Ser310Ala) | 49 | rs6691840 | 0.2747 | 0.30611 | 0.0455 | Benign | Tolerated | Neutral | Neutral | 0.54/9.832 |
| chr1:37325477 | c.928T > C (p.Ser310Pro) | 2 | rs6691840 | 0.0001 | N/R | 0.0017 | Benign | Tolerated | Neutral | Neutral | 0.60/10.34 |
| chr1:37324735 | c.1078G > A (p.Gly360Ser) | 2 | rs551525926 | 0.0002 | 0.0004 | 0.0049 | Benign | Tolerated | Neutral | Neutral | 2.79/23.1 |
| chr1:37271783 | c.2236G > A (p.Val746Ile) | 1 | rs142411639 | <0.0001 | N/R | 0.0003 | Benign | Tolerated | Neutral | Neutral | 1.41/15.77 |
| chr1:37267613 | c.2599C > T (p.Arg867Cys) | 2 | rs758481140 | <0.0001 | N/R | 0.0003 | Probably damaging | Tolerated | Neutral | Deleterious | 3.26/24.2 |
| chr1:37267505 | c.2707C > G (p.Pro903Ala) | 1 | rs777066849 | <0.0001 | N/R | 0.0010 | Benign | Damaging | Neutral | Neutral | 2.96/23.5 |
| GRIK4 | |||||||||||
| chr11:120702714 | c.665C > G (p.Ser222Cys) | 1 | rs747558889 | <0.0001 | N/R | 0.0004 | Benign | Damaging | Neutral | Deleterious | 3.06/23.7 |
| chr11:120744892 | c.1024C > T (p.Gln342Ter) | 1 | N/R | N/R | N/R | N/R | N/A | N/A | N/A | N/A | N/A |
| chr11:120769281 | c.1205G > A (p.Ser402Asn) | 1 | rs776598317 | <0.0001 | N/R | N/R | Benign | Tolerated | Neutral | Neutral | 1.82/18.37 |
| chr11:120769285 | c.1209C > A (p.His403Gln) | 1 | N/R | N/R | N/R | N/R | Benign | Tolerated | Neutral | Neutral | 0.89/12.66 |
| chr11:120776105 | c.1379G > A (p.Arg460Gln) | 1 | rs777568733 | <0.0001 | N/R | 0.0003 | Benign | Damaging | Neutral | Neutral | 2.16/21.1 |
| chr11:120811098 | c.1519C > T (p.Arg507Trp) | 1 | rs747648319 | <0.0001 | N/R | 0.0003 | Probably damaging | Damaging | Disease | Deleterious | 4.41/29.0 |
| chr11:120827536 | c.1748G > A (p.Arg583Gln) | 1 | rs367593579 | <0.0001 | N/R | 0.0003 | Probably damaging | Tolerated | Neutral | Neutral | 3.79/26.2 |
| chr11:120827587 | c.1799C > T (p.Pro600Leu) | 1 | rs766921998 | <0.0001 | N/R | N/R | Probably damaging | Tolerated | Disease | Deleterious | 4.16/29.2 |
| chr11:120837916 | c.2279G > C (p.Arg760Pro) | 1 | rs199658262 | <0.0001 | 0.0002 | N/R | Probably damaging | Damaging | Disease | Deleterious | 4.42/32 |
| chr11:120856667 | c.2569G > A (p.Asp857Asn) | 8 | rs536558955 | 0.0004 | 0.0008 | 0.0100 | Benign | Tolerated | Neutral | Neutral | 2.87/23.3 |
| chr11:120856715 | c.2617C > T (p.Pro873Ser) | 3 | rs1485463780 | <0.0001 | N/R | 0.0005 | Benign | Tolerated | Neutral | Neutral | 1.92/19.14 |
| chr11:120856865 | c.2767C > A (p.Pro923Thr) | 1 | rs772831685 | N/R | N/R | 0.0005 | Possibly damaging | Tolerated | Neutral | Neutral | 2.87/23.3 |
| chr11:120856878 | c.2780G > A (p.Arg927His) | 2 | rs537835045 | <0.0001 | 0.0002 | 0.0015 | Probably damaging | Damaging | Neutral | Neutral | 4.45/32 |
| GRIK5 | |||||||||||
| chr19:42569506 | c.113G > A (p.Arg38His) | 3 | rs143068269 | 0.0004 | 0.0004 | N/R | Probably damaging | Damaging | Neutral | Neutral | 4.13/28.9 |
| chr19:42563662 | c.526G > A (p.Glu176Lys) | 3 | rs201724297 | 0.0002 | 0.0010 | 0.0030 | Benign | Tolerated | Neutral | Neutral | 3.04/23.7 |
| chr19:42561126 | c.692C > T (p.Ser231Leu) | 1 | rs557315821 | <0.0001 | 0.0002 | 0.0023 | Probably damaging | Tolerated | Neutral | Neutral | 2.83/23.2 |
| chr19:42558591 | c.937G > A (p.Val313Met) | 8 | rs746539777 | <0.0001 | N/R | 0.0023 | Probably damaging | Tolerated | Neutral | Neutral | 3.18/24.0 |
| chr19:42557839 | c.1184G > A (p.Arg395His) | 2 | rs138759162 | 0.0003 | 0.0006 | 0.0010 | Possibly damaging | Tolerated | Neutral | Neutral | 2.97/23.5 |
| chr19:42557801 | c.1222C > G (p.Leu408Val) | 1 | rs749428580 | <0.0001 | N/R | N/R | Benign | Tolerated | Neutral | Neutral | 1.33/15.37 |
| chr19:42525493 | c.1831A > T (p.Met611Leu) | 1 | rs772424091 | <0.0001 | N/R | 0.0010 | Benign | Tolerated | Neutral | Neutral | 2.27/21.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, T.-M.; Wu, C.-L.; Hsu, S.-H.; Tsai, H.-Y.; Cheng, F.-Y.; Cheng, M.-C. Ultrarare Loss-of-Function Mutations in the Genes Encoding the Ionotropic Glutamate Receptors of Kainate Subtypes Associated with Schizophrenia Disrupt the Interaction with PSD95. J. Pers. Med. 2022, 12, 783. https://doi.org/10.3390/jpm12050783
Hu T-M, Wu C-L, Hsu S-H, Tsai H-Y, Cheng F-Y, Cheng M-C. Ultrarare Loss-of-Function Mutations in the Genes Encoding the Ionotropic Glutamate Receptors of Kainate Subtypes Associated with Schizophrenia Disrupt the Interaction with PSD95. Journal of Personalized Medicine. 2022; 12(5):783. https://doi.org/10.3390/jpm12050783
Chicago/Turabian StyleHu, Tsung-Ming, Chia-Liang Wu, Shih-Hsin Hsu, Hsin-Yao Tsai, Fu-Yu Cheng, and Min-Chih Cheng. 2022. "Ultrarare Loss-of-Function Mutations in the Genes Encoding the Ionotropic Glutamate Receptors of Kainate Subtypes Associated with Schizophrenia Disrupt the Interaction with PSD95" Journal of Personalized Medicine 12, no. 5: 783. https://doi.org/10.3390/jpm12050783
APA StyleHu, T. -M., Wu, C. -L., Hsu, S. -H., Tsai, H. -Y., Cheng, F. -Y., & Cheng, M. -C. (2022). Ultrarare Loss-of-Function Mutations in the Genes Encoding the Ionotropic Glutamate Receptors of Kainate Subtypes Associated with Schizophrenia Disrupt the Interaction with PSD95. Journal of Personalized Medicine, 12(5), 783. https://doi.org/10.3390/jpm12050783