1. Introduction
The technologies nowadays available to reduce friction and wear are based on materials, an important class of which is represented by lubricant additives included in motor oils. The design of lubricant additives to decrease friction and wear in machine components is an important way to increase the energy efficiency of car engines while taking into account restrictive environmental requirements and technological advances [
1]. Lubricants are formulated products composed of base oils and a package of additives designed for specific performance needs. The additives can be classified as chemically active, i.e., designed to chemically interact with the surface and form protective layers; or chemically inert, i.e., with the function of improving the physical properties of the base oil. Most of the chemically active additives have a non-polar part, usually consisting of a hydrocarbon chains that solubilize the molecule into the base oil, and a functional polar group that is tailored for the specific function that the additive should carry out. Here we consider extreme-pressure (EP) additives that operate in boundary lubrication conditions, where the thickness of the oil film becomes too thin to prevent direct contact between the metal asperities. EP additives typically adsorb onto the metal surface either by physical or chemical attraction [
2,
3]. Under severe tribological conditions, they react with the surfaces forming surface films that prevent the welding of opposing asperities and avoid scuffing that is destructive to sliding surfaces under high loads [
4]. To improve boundary lubricants, it is highly desirable to understand how these protective films are formed by tribochemical reactions. However, many aspects of tribochemistry remain elusive due to the difficulties in directly probing the buried interface. From post mortem spectroscopy, is possible to acquire information on the reaction products, but very little can be inferred on the activation mechanisms, reaction pathways, and rates. Simulations can play a decisive role here [
5,
6], in particular ab initio molecular dynamics (AIMD), where both the ionic and electronics degrees of freedom are fully taken in into account. This is essential for an accurate description of bond-breaking and bond-forming reactions in the situation of enhanced reactivity imposed by the boundary lubrication conditions. However, the use of AIMD in tribology has been traditionally very scarce and most of the existing simulations of tribochemistry are based on force fields, i.e., on a parameterization of the atomic interactions. While many force fields are available for selected chemical environments, their transferability is often poor, especially to describe the non-ordinary conditions present at the tribological interface.
Here we apply AIMD to investigate tribochemistry mechanisms that govern the functionality of EP additives where the key element of the functional group is phosphorus. P-based compounds are largely used as EP and anti-wear (AW) additives for gear oils [
7,
8,
9]. The chemical structure of P-containing additives influences the nature and the tribological performances of the tribofilm as highlighted in [
10,
11,
12], where phosphite and phosphate have been compared. Typically, organic phosphites function as friction-modifiers, whereas phosphates function as AW additives.
A convenient experimental set-up to study the tribochemistry of lubricant additives in boundary lubrication conditions is represented by gas phase lubrication (GPL), where gaseous molecules containing the same functional group of commercial additives are introduced in the tribometer chamber and react with the metallic sliding parts in contact in the absence of the base oil. Among all types of phosphite additives, trimethylphosphite (TMPi), with chemical formulae P(CH
3O)
3, is probably the simplest compound containing the phosphite ion, thus it was chosen in the GPL experiments performed by Phillipon et al. to provide a basic understanding on the functionality of organophosphorous additives [
13,
14,
15]. The experiment revealed that TMPi decomposes on nascent iron surfaces and the tribological conditions favor the formation of an iron phosphide tribofilm that significantly reduces the coefficient of friction and wear at the steel–steel sliding contact.
To identify the mechanisms of TMPi dissociation, we combined first principles calculation and XPS analysis of TMPi adsorption and dissociation at the open Fe(110) surface [
16]. The calculated reaction energies indicate that dissociation is energetically more favorable than molecular adsorption and it is a thermally activated process. In situ XPS analysis of adsorbed TMPi on metallic iron confirmed molecular chemisorption and dissociation at high temperature. In the present paper, the effects of tribological conditions, which include confinement, mechanical stresses (load and shear) on the reaction of TMPi dissociation are highlighted. This is accomplished by comparing the results obtained at the open surface in static conditions with AIMD imitations of TMPi molecules confined at sliding iron interfaces under load. These AIMD simulations represent a great computational challenge due to the difficulty imposed by the presence of a metallic system where the ionic and electronic degrees of freedom can exchange energy due to the absence of a band gap. In addition, the magnetic character of iron imposes the use of spin polarization. We face this challenge by adopting the Born–Oppenheimer (BO) scheme, where the ions are moved according to the Hellmann–Feynman forces obtained from the total electronic energy, which is minimized every MD step. In this way, the ionic and electronic degrees of freedom are decoupled and the dynamics of metallic systems, even including magnetization, can be described in accurate way. The observed tribochemical reactions clarify the mechanism of P release at the buried interface, in agreement with the experimental observations and with the first-principle study of molecular decomposition at the open surface [
16]. However, the observed dissociation rates are much higher in tribological conditions: By repeating the tribological AIMD simulation at different applied loads, we highlight the primary role of mechanical stresses in promoting the reactions.
2. Materials and Methods
We perform spin-polarized density functional theory (DFT) calculations, using a modified version of the BO MD code included in the Quantum Espresso package (version 5.0.3) [
17]. The exchange–correlation functional is described by the Perdew–Burke–Ernzerhof (PBE) parameterization [
18]. The electronic wave functions are expanded in plane-waves with kinetic energy cut-off of 30 Ry (240 Ry for the charge density). The Fe(110) surface is considered since it is the most stable among the densely packed iron surfaces [
19]. The surface is modeled by means of periodic supercells containing an iron slab of 4 × 4 in-plane size, e.g., 16 atoms per layer, and a vacuum region 20 Å thick. It has been verified that the choice of the 4 × 4 in-plane size is sufficient to avoid lateral interaction of TMPi with its periodic replicas. The Brillouin zone sampling is realized by means of a 2 × 2 × 1 Monkhorst Pack grid [
20]. Iron interfaces, are modeled by two self-mated Fe(110) surfaces. During the AIMD simulations the bottom layer of the lower slab (the substrate) was held rigid, while the upper slab (the counter-surface) was moved at constant velocity of 200 m/s. This velocity is much higher than the typical sliding speed in a tribometer at boundary conditions (~70 mm/s). However, the agreement between the reaction path observed in the AIMD simulation and that predicted by static first principles calculations, which are also consistent with thermal decomposition observed in experiments, lead us to believe that the high sliding velocity imposed in the dynamic simulation does not alter the reaction paths of the chemical processes. In other words, the chemical processes that are most likely to occur on the basis of thermodynamics are the same observed at high sliding velocity. A vertical force was applied to the atoms belonging to the top layer of the counter-surface to model an applied pressure the value of which was varied from 500 MPa to 5 GPa to evaluate the dependence of the reaction rates on load. In the initial configuration, common to all the performed simulations, a TMPi molecule per cell (of ~100 Å
2 area) was adsorbed in its most stable configuration on the iron substrate and the system was relaxed under the effects of the applied pressure. After the relaxation at 500 MPa pressure, the system was equilibrated at a constant temperature of 300 K. This starting system configuration is common to all the simulations at different applied loads.
3. Results
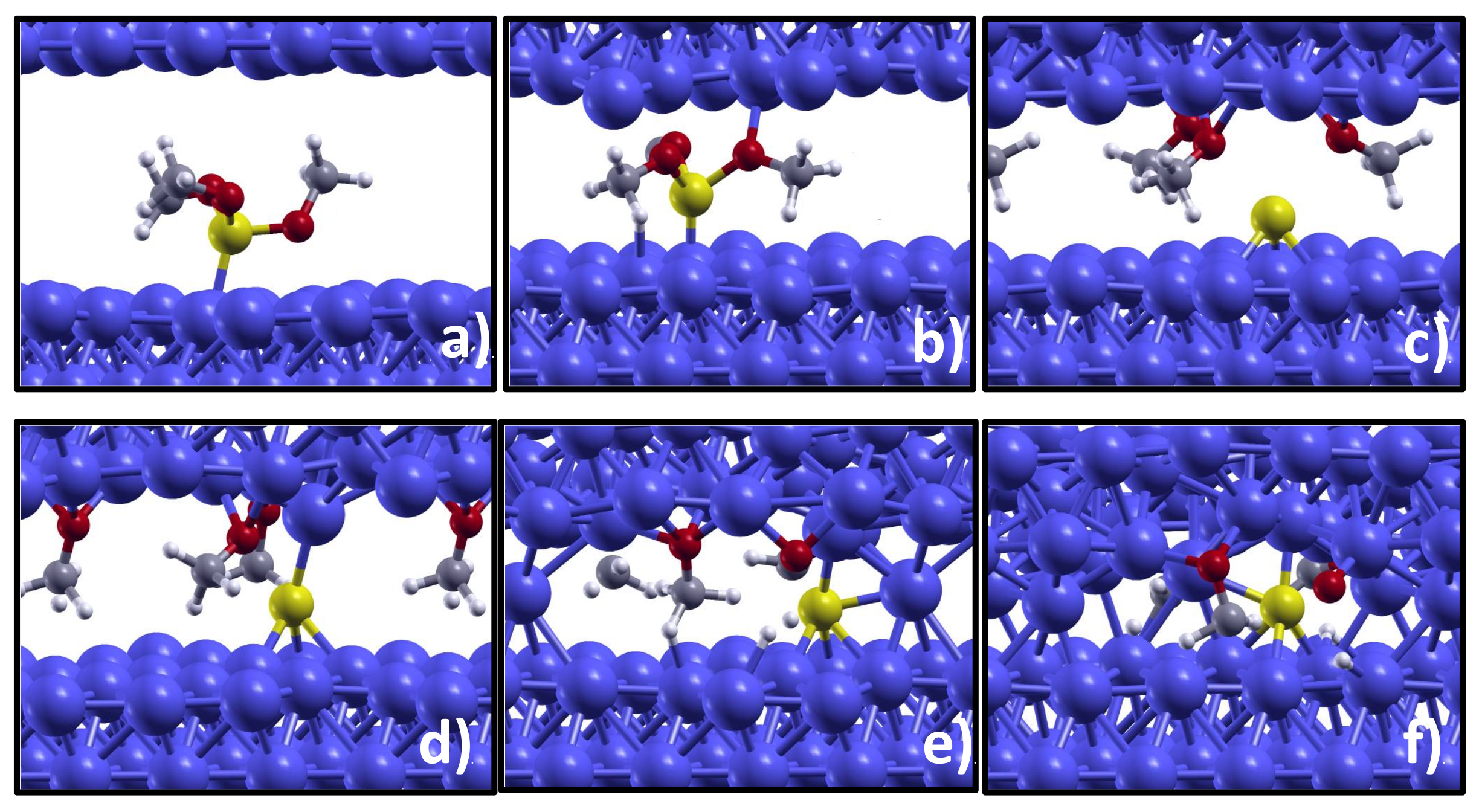
The first AIMD simulation, which is carried out at 500 MPa load, is available as
Video S1 in the Supplemental Information (SI). The system dynamics observed during a time interval of 7 ps consists in molecular vibrations, mainly rotations of each methyl group around the axis along the CO bond, and we do not observe any dissociative reaction. By increasing the pressure to 2 GPa, molecular dissociation is, instead, observed after 1 ps. As can be seen in
Figure 1a,b, the increased load makes the iron counter-surface to get closer to the substrate. This load-induced confinement promotes the molecular dissociation that occurs via PO bond breaking. As can be seen in the atom trajectory reported as
Video S2, the oxygen atoms are attracted by the incoming iron counter-surface, the PO bonds get stretched and, finally, methoxy groups (OCH
3) dissociate and adsorb on the counter-surface leaving elemental phosphorus remains on the substrate (
Figure 1c). We notice that the P atom is located at a long bridge site, which is indicated as the most favorable site for P adsorption at the Fe(110) surface by first principles calculations of adatom chemisorption [
21].
The simulation snapshot presented in panel (d) highlights the tendency of phosphorus at low concentration to interact with the Fe counter-surface: a Fe atom not involved in FeO bonds is ‘captured’ by the P atom and drags downward the substrate. The surface separation is reduced and a direct contact between few metal atoms established (
Figure 1e). It is very interesting to notice that this direct metal–metal contact is sufficient to promote the cold sealing of the two iron surfaces. Upon surface sealing, the molecular fragments remain embedded in the iron matrix (
Figure 1f). This event in real life would be detrimental, corresponding e.g., to engine seizure. However, we expect that the direct metal–metal contact can be prevented by an increased phosphorous coverage. This would explain the wear and friction reduction provided by the iron phosphide tribofilm, observed in the experiments. In
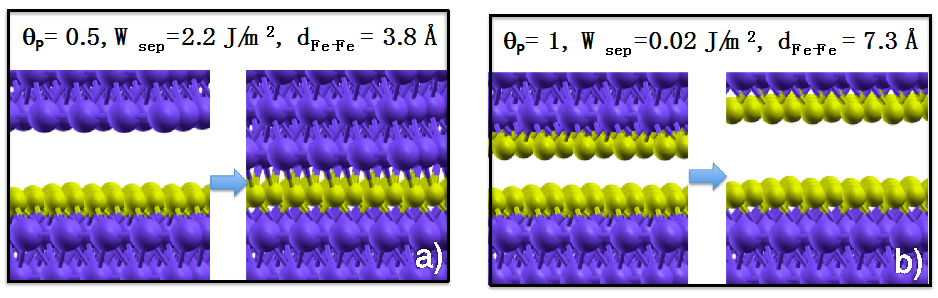
Figure 2, we show the initial and final configurations of the optimization process at zero applied load for an iron interface passivated with 0.5 ML (a) and 1 ML of P atoms (b). We can observe that the interfacial P highly reduces the work of separation of iron (
Wsep of clean iron is 4.8 J/m
2). Moreover, direct Fe–Fe contact is inhibited at the interface at the considered P coverages: At 0.5 ML coverage chemical Fe–P–Fe bonds are present across the interface, while at 1 ML coverage the fully passivated surfaces repulse each other and the Fe–Fe distance increases to 7.3 Å. The observed adhesion reduction is accompanied by a dramatic decrease of the interfacial shear strength [
21].
In the AIMD simulation at the higher applied load of 5 GPa the molecular dissociation occurs following the same path as observed at 2 GPa, but the dissociation takes place just 0.6 ps after the beginning of the simulation (
Video S3 of SI). This suggests that the activation rate of tribochemical reactions increases with the load applied at the sliding interface, as discussed in the next session.
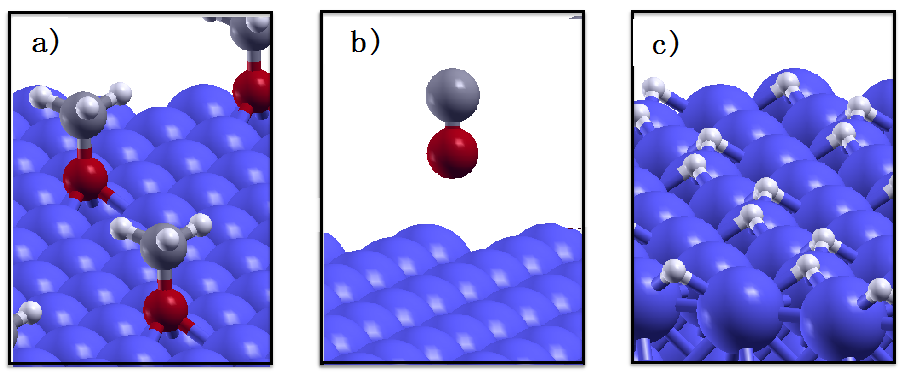
A closer inspection of the molecular fragments embedded in the iron matrix (
Figure 1f) reveals that the methoxy groups got dissociated into H atoms on CO molecules. To identify the driving force for this process, we calculate the reaction energy,
ER, associated to methoxy dehydrogenation. First identify the most favorable adsorption sites for the OCH
3 group, CO, and H on the Fe(110) surface. The reaction energy is, in fact, calculated as the difference between the adsorption energy of the methoxy group and that of the adsorbed CO and H fragments. We find that the most favorable adsorption site for both methoxy and H adsorption is the three-fold site, where the adsorbates are bounded with three Fe atoms (
Figure 3a,c). The CO molecule, instead, does not chemisorb on iron: during the relaxation process the molecule detaches from the surface and remains physisorbed at 4.1 Å distance from the on-top site, where physisorption is more favorable (
Figure 3b). By tilting the molecule upside-down with the C atom closer to the surface we obtain a slightly lower physisorption attraction. The calculated reaction energy for methoxy dehydrogenation is −0.87 eV. The negative sign indicates that this process is energetically favored, confirming the reliability of the AIMD simulation.
4. Discussion
The reaction path observed during the AIMD simulation indicates that the molecular dissociation occurs through the cleavage of the PO bonds that cause methoxy detachment. As can be seen by comparing
Figure 1a,b, the O atoms of the molecule are attracted by the Fe counter-surface and when the latter is close enough, the methoxy groups detach from the TMPi molecule and adsorb on it. These findings indicate that clean Fe sites are necessary for TMPi dissociation on the surface and explain the important role of the nascent surfaces in favoring the adsorption and dissociation of TMPi observed experimentally [
13]. Particularly, the freshly scratched surfaces in a vacuum might expose a higher number of Fe atoms than flat surfaces.
The decomposition path highlighted by the AIMD simulations is perfectly consistent with the decomposition path identified by combining temperature-programmed reaction spectroscopy, X-ray photoelectron spectroscopy (XPS), and low-energy electron diffraction. In this experiment, it is observed that TMPi decomposes on the clean Fe(110) into adsorbed phosphorous, gaseous CO, and H
2 via the methoxy intermediate, leaving behind a phosphide film [
22]. The thermally-activated formation of iron phosphide has been also observed upon TMPi adsorption at etched steel surfaces. The characteristic peak of iron phosphide appeared clearly at a temperature of 300 °C. These results strongly suggest that the formation of iron phosphide occurs if an energy barrier is overcome [
21].
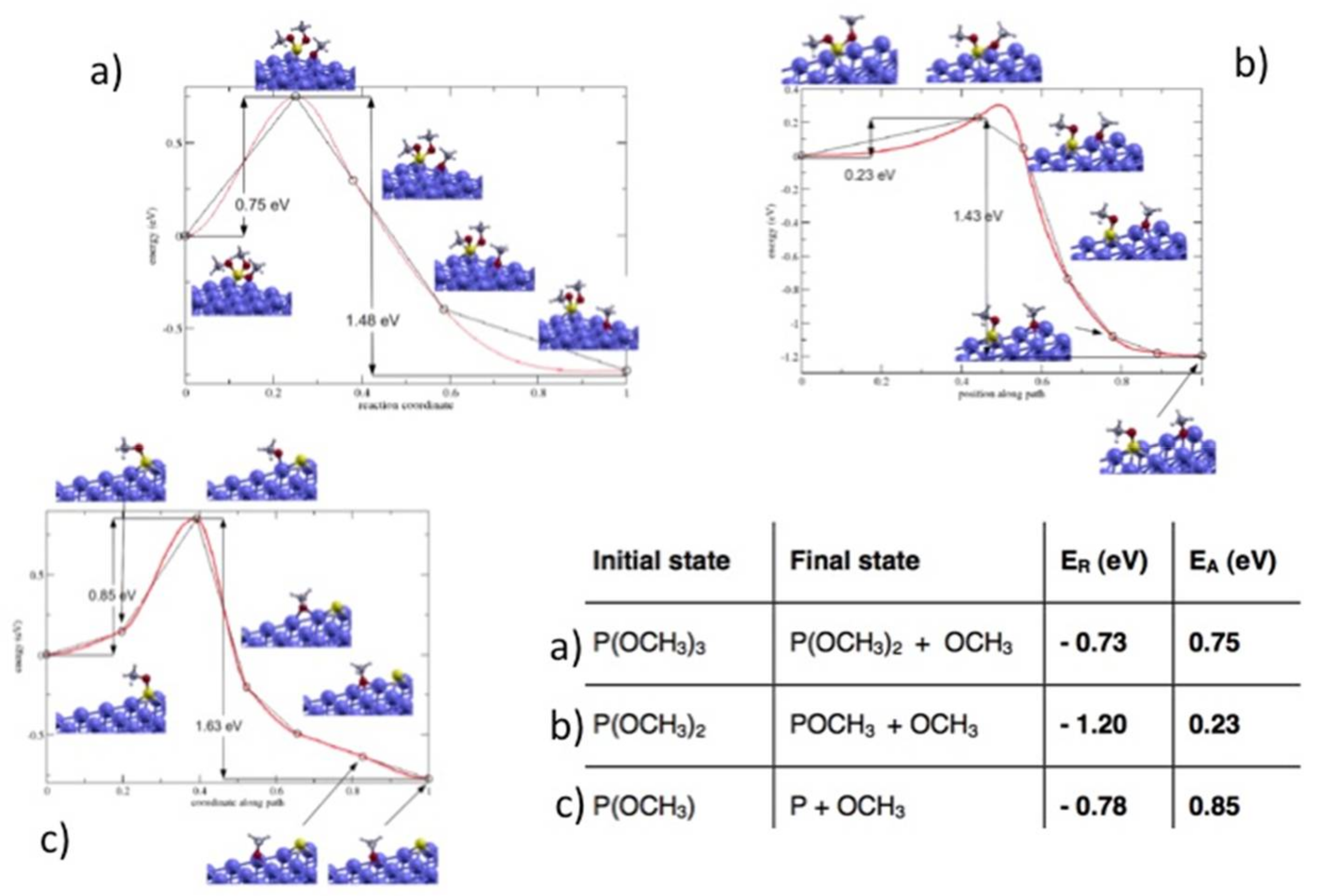
We estimated the height of the energy barrier for TMPi dissociation at the Fe(110) surface by first principles calculations [
21]. The results obtained by applying the nudged elastic band method [
23] are schematically represented in
Figure 4. The three panels describe the calculated reaction paths for the subsequent detachment of the methoxy groups, while the corresponding reaction energies and barriers are reported in the table enclosed in the picture. The negative values of
ER indicate that the molecular dissociation via methoxy detachment is a highly exothermic process and the energy gain increases until the full molecular decomposition in atomic P and 3 adsorbed methoxy groups is realized. It is interesting to notice that these thermodynamical driving forces can explain the molecular dissociation observed during the dynamic simulation.
The calculated activation barriers,
EA, indicate that methoxy detachment occurs upon energy transfer, as observed in the thermal programmed experiments above described. The fact that the TMPi dissociation is observed in our AIMD simulations at room temperature highlights the capability of mechanical stresses to activate the reaction. Moreover, the activation time observed in the tribological simulation turned out to be much shorter than the activation time expected by applying the Arrhenius law at room temperature, considering a limiting barrier of 0.75–0.85 eV, (Tab of
Figure 4). This result, along with the trend that we observed by increasing the applied load from 500 MPa to 2 and 5 GPa indicates that the load-induced confinement plays a crucial role in accelerating the reaction rates. The important role of load-induced confinement has been previously highlighted by AIMD simulations of tribochemical reactions involving water molecules confined at diamond interfaces [
24,
25]. In particular, it was found that the higher the load, the more effective the surface passivation by water dissociation, which in turn leads to a reduction of friction [
24]. AIMD simulations offer the opportunity to isolate the effects of mechanical stresses from other effects which have been proposed to activate tribochemical reactions. The first proposed mechanisms [
26] attributed the increase of the reaction rates to extremely high temperatures, “flash temperatures” that develop a few discrete “hot spots”, where the contact between the two surfaces occurs [
27]. Such temperature increases have also been proposed to produce local excitations, “magma plasma”, that decay rapidly into a local heating of the surface. However, recent works have shown that tribochemical reactions can take place even though the temperature raise during sliding is negligible or limited and highlighted the primary role of mechanical stresses in the activation of chemical reactions [
28,
29,
30,
31,
32,
33,
34,
35,
36,
37].
A second important outcome of the tribological AIMD simulations concerns the reaction products. As observed under extreme confinement, the final products of TMPi dissociation are CO molecules and atomic P. This simple observation accompanied by the evidence we provide on the effects of P in reducing the adhesion and increasing the metal separation is sufficient to uncover the mechanisms underlying the friction reduction observed in GPL by TMPi. Our results show, in fact, that in boundary lubrication conditions, elemental O and C atoms are not released from TMPi because these two atoms remain bounded in the CO molecule, which does not dissociate on iron [
38] and can desorb from the interface. H atoms can desorb as well in the form of H
2 or diffuse into the iron bulk. Therefore, what remains at the interface after TMPi decomposition is elemental P, as clearly identified by XPS analysis performed in situ after the GPL experiment [
16]. Iron phosphide presents low friction, as evidenced by the experiment and in first principles calculations [
21]. While a similar lubricating effect can be provided by sulfur [
39] and graphene [
40,
41,
42], it is not provided by atomic oxygen [
21]. The AIMD simulations reveal that O adsorption does not compete with P adsorption during TMPi decomposition. This explains the higher efficiency of phosphites than phosphates, where an extra oxygen per molecule is present, in reducing friction observed in the experiments [
21]. It is important to notice that the presence of iron oxide on the surface could alter the dissociation path, e.g., in aryl phosphates and phosphites there is evidence that the C–O bond cleavage is more likely than the P–O bond.
A recent first principles study of P chemisorption described in a forthcoming article revealed that by increasing the P coverage on the Fe(110) surface, the lateral interaction among the adsorbates increases. This in-plane interaction is responsible for the decrease of adhesion observed when two P-covered surfaces are brought into contact. The next step in our analysis of the tribochemistry of organophosfurus additives will be to consider a larger number of additive molecules in our supercell to monitor the formation and effects of iron phosphide at higher concentrations of P in real time. Such simulation requires enlarging the size of the simulated system, with consequent increase of the computational cost of AIMD. This computational challenge can be face e.g., by hybrid quantum-mechanics/molecular-mechanics (QM/MM) technique, recently applied for the first time to tribology by our group.



{kind=link}
{kind=link}
{kind=link}
{kind=link}



