
Annotating the Insect Regulatory Genome


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Empirical Approaches to CRM Discovery
2.1. Reporter Gene Assays
2.2. ChIP-Based Assays
2.3. Limitations
3. Computational Approaches
3.1. Supervised Machine Learning
3.2. Chromatin and Epigenetic Features
3.3. Sequence Features
4. Cross-Species or Non-Model Insect CRM Discovery
5. REDfly and SCRMshaw: Powerful Tools for Insect Regulatory Genomics
5.1. REDfly
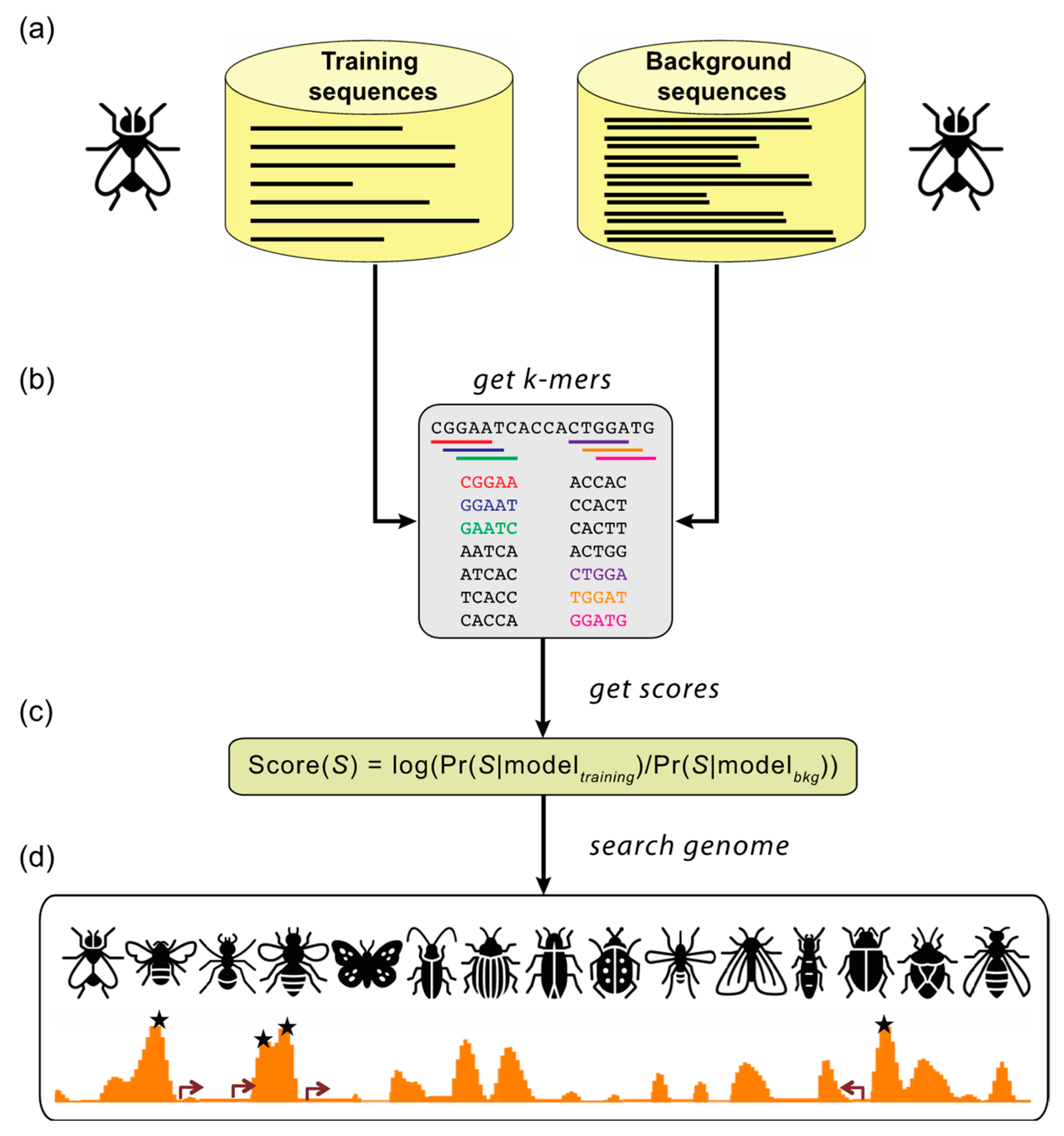
5.2. SCRMshaw
5.3. Cross-Species Prediction
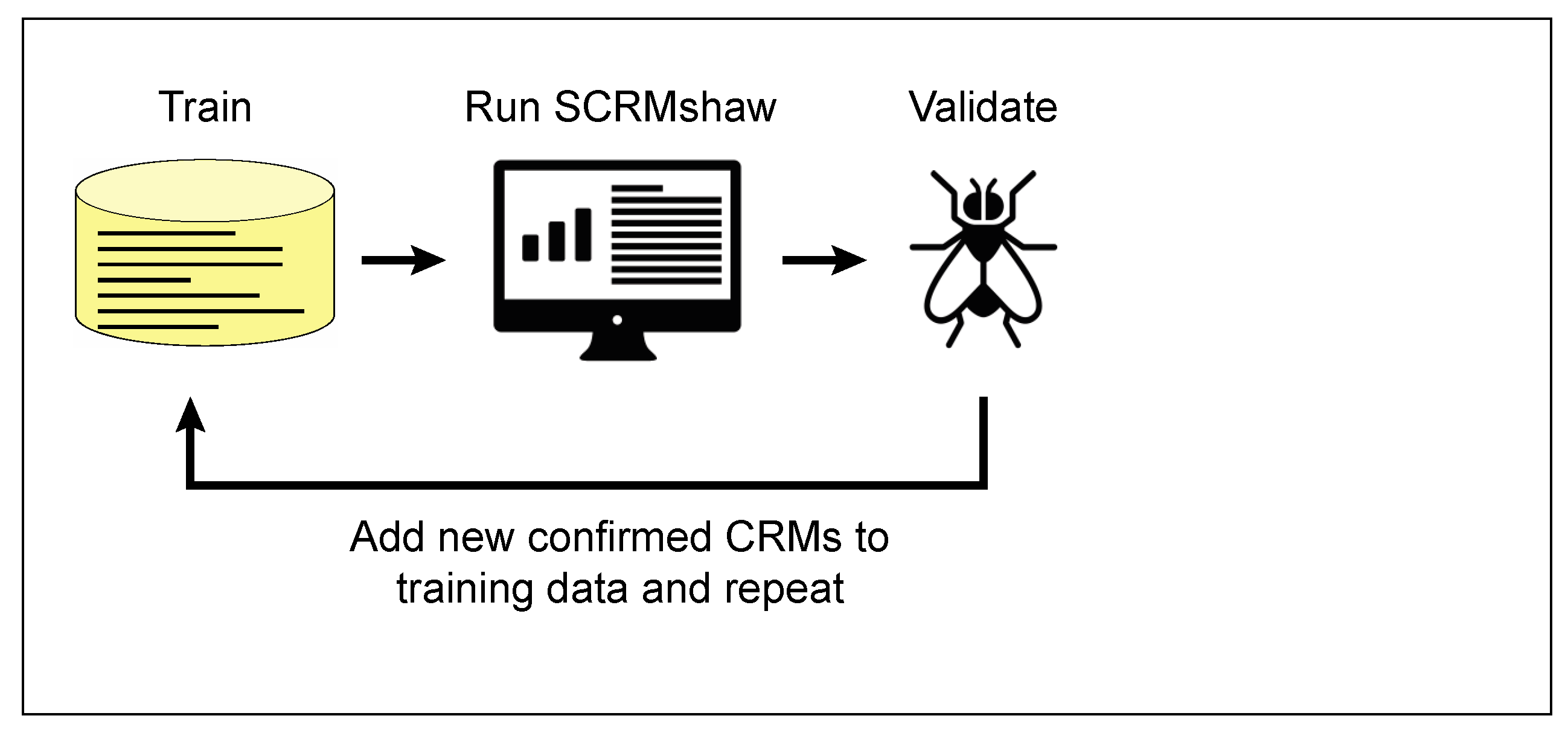
5.4. Training Set Improvement
5.5. Limitations
6. Integrating Computational and Experimental Approaches
7. Conclusions
8. URLs
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANN | Artificial neural network |
| ATAC-seq | Assay for transposase-accessible chromatin using sequencing |
| auROC | Area under the receiver operating characteristic |
| BRNN | Bidirectional recurrent neural networks |
| ChIA-PET | Chromatin interaction analysis with paired-end tag |
| ChIP | Chromatin immuno precipitation |
| CNN | Convolutional neural network |
| CRM | cis-regulatory module |
| CSI-ANN | Chromatin signature identification by artificial neural network |
| DECRES | Deep learning for identifying cis-regulatory elements |
| ECNN | Ensemble of CNN |
| ENCODE | Encyclopedia of DNA elements |
| FAIRE | Formaldehyde-assisted isolation of regulatory elements |
| FANTOM | Functional annotation of the mammalian genome |
| Gkm-SVM | Gapped kmer support vector machine |
| HMM | Hidden Markov model |
| Kmer-SVM | kmer-support vector machine |
| ML | Machine learning |
| REDfly | Regulatory element database for Drosophila |
| RF | Random forest |
| RFECS | Random forest-based enhancer identification using chromatin states |
| ROC | Receiver operating characteristic |
| scATAC-seq | Single cell assay for transposase-accessible chromatin using sequencing |
| SCRMshaw | Supervised cis-regulatory module discovery |
| STARR-seq | Self-transcribing active regulatory region sequencing |
| SVM | Support vector machine |
| TF | Transcription factor |
| TFBS | Transcription factor binding site |
References
- NCBI Genome Information by Organism. Available online: https://www.ncbi.nlm.nih.gov/genome/browse#!/overview/ (accessed on 25 May 2021).
- i5K Consortium. The i5K Initiative: Advancing arthropod genomics for knowledge, human health, agriculture, and the environment. J. Hered. 2013, 104, 595–600. [Google Scholar] [CrossRef]
- Davidson, E.H.; Erwin, D.H. Gene regulatory networks and the evolution of animal body plans. Science 2006, 311, 796–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, S.B.; Grenier, J.K.; Weatherbee, S.D. From DNA to Diversity. Molecular Genetics and the Evolution of Animal Design; Blackwell Science: Malden, MA, USA, 2001. [Google Scholar]
- Pennacchio, L.A.; Bickmore, W.; Dean, A.; Nobrega, M.A.; Bejerano, G. Enhancers: Five essential questions. Nat. Rev. Genet. 2013, 14, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Halfon, M.S. Studying Transcriptional Enhancers: The Founder Fallacy, Validation Creep, and Other Biases. Trends Genet. 2019, 35, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Catarino, R.R.; Stark, A. Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev. 2018, 32, 202–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasperini, M.; Tome, J.M.; Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 2020, 21, 292–310. [Google Scholar] [CrossRef]
- Rivera, J.; Keranen, S.V.E.; Gallo, S.M.; Halfon, M.S. REDfly: The transcriptional regulatory element database for Drosophila. Nucleic Acids Res. 2019, 47, D828–D834. [Google Scholar] [CrossRef] [Green Version]
- Kantorovitz, M.R.; Kazemian, M.; Kinston, S.; Miranda-Saavedra, D.; Zhu, Q.; Robinson, G.E.; Göttgens, B.; Halfon, M.S.; Sinha, S. Motif-blind, genome-wide discovery of cis-regulatory modules in Drosophila and mouse. Dev. Cell 2009, 17, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Kazemian, M.; Zhu, Q.; Halfon, M.S.; Sinha, S. Improved accuracy of supervised CRM discovery with interpolated Markov models and cross-species comparison. Nucleic Acids Res. 2011, 39, 9463–9472. [Google Scholar] [CrossRef] [Green Version]
- Kazemian, M.; Halfon, M.S. CRM Discovery Beyond Model Insects. In Insect Genomics: Methods and Protocols; Brown, S.J., Pfrender, M.E., Eds.; Springer: New York, NY, USA, 2019; pp. 117–139. [Google Scholar]
- Kwasnieski, J.C.; Mogno, I.; Myers, C.A.; Corbo, J.C.; Cohen, B.A. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc. Natl. Acad. Sci. USA 2012, 109, 19498–19503. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryn, L.M.; Rath, M.; Stark, A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef]
- Murtha, M.; Tokcaer-Keskin, Z.; Tang, Z.; Strino, F.; Chen, X.; Wang, Y.; Xi, X.; Basilico, C.; Brown, S.; Bonneau, R.; et al. FIREWACh: High-throughput functional detection of transcriptional regulatory modules in mammalian cells. Nat. Methods 2014, 11, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Johnson, G.D.; Seo, J.; Barrera, A.; Cowart, T.N.; Majoros, W.H.; Ochoa, A.; Allen, A.S.; Reddy, T.E. Correcting signal biases and detecting regulatory elements in STARR-seq data. Genome Res. 2021, 31, 877–889. [Google Scholar] [CrossRef]
- Lee, D.; Shi, M.; Moran, J.; Wall, M.; Zhang, J.; Liu, J.; Fitzgerald, D.; Kyono, Y.; Ma, L.; White, K.P.; et al. STARRPeaker: Uniform processing and accurate identification of STARR-seq active regions. Genome Biol. 2020, 21, 298. [Google Scholar] [CrossRef]
- Peng, T.; Zhai, Y.; Atlasi, Y.; Ter Huurne, M.; Marks, H.; Stunnenberg, H.G.; Megchelenbrink, W. STARR-seq identifies active, chromatin-masked, and dormant enhancers in pluripotent mouse embryonic stem cells. Genome Biol. 2020, 21, 243. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M. Shooting for the STARRs: A Modified STARR-seq Assay for Rapid Identification and Evaluation of Plant Regulatory Sequences in Tobacco Leaves. Plant Cell 2020, 32, 2057–2058. [Google Scholar] [CrossRef]
- Zhang, J.; Lee, D.; Dhiman, V.; Jiang, P.; Xu, J.; McGillivray, P.; Yang, H.; Liu, J.; Meyerson, W.; Clarke, D.; et al. An integrative ENCODE resource for cancer genomics. Nat. Commun. 2020, 11, 3696. [Google Scholar] [CrossRef] [PubMed]
- Asma, H.; Halfon, M.S. Computational enhancer prediction: Evaluation and improvements. BMC Bioinform. 2019, 20, 174. [Google Scholar] [CrossRef] [Green Version]
- Ghavi-Helm, Y.; Furlong, E.E. Analyzing transcription factor occupancy during embryo development using ChIP-seq. Methods Mol. Biol. 2012, 786, 229–245. [Google Scholar] [CrossRef]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Bell, O.; Tiwari, V.K.; Thoma, N.H.; Schubeler, D. Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 2011, 12, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, D.J. Using Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE) to Identify Functional Regulatory DNA in Insect Genomes. Methods Mol. Biol. 2019, 1858, 89–97. [Google Scholar] [CrossRef]
- McKay, D.J.; Lieb, J.D. A common set of DNA regulatory elements shapes Drosophila appendages. Dev. Cell 2013, 27, 306–318. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.21–21.29.29. [Google Scholar] [CrossRef]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Bravo Gonzalez-Blas, C.; Quan, X.J.; Duran-Romana, R.; Taskiran, I.I.; Koldere, D.; Davie, K.; Christiaens, V.; Makhzami, S.; Hulselmans, G.; de Waegeneer, M.; et al. Identification of genomic enhancers through spatial integration of single-cell transcriptomics and epigenomics. Mol. Syst. Biol. 2020, 16, e9438. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef]
- Chen, X.; Miragaia, R.J.; Natarajan, K.N.; Teichmann, S.A. A rapid and robust method for single cell chromatin accessibility profiling. Nat. Commun. 2018, 9, 5345. [Google Scholar] [CrossRef] [PubMed]
- Mezger, A.; Klemm, S.; Mann, I.; Brower, K.; Mir, A.; Bostick, M.; Farmer, A.; Fordyce, P.; Linnarsson, S.; Greenleaf, W. High-throughput chromatin accessibility profiling at single-cell resolution. Nat. Commun. 2018, 9, 3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.; Lee, I. Single-cell ATAC sequencing analysis: From data preprocessing to hypothesis generation. Comput. Struct. Biotechnol. J. 2020, 18, 1429–1439. [Google Scholar] [CrossRef]
- Fiers, M.; Minnoye, L.; Aibar, S.; Bravo Gonzalez-Blas, C.; Kalender Atak, Z.; Aerts, S. Mapping gene regulatory networks from single-cell omics data. Brief Funct. Genom. 2018, 17, 246–254. [Google Scholar] [CrossRef]
- Blow, M.J.; McCulley, D.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 2010, 42, 806–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, D.; Blow, M.J.; Kaplan, T.; McCulley, D.J.; Jensen, B.C.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 2011, 44, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, M.; Miranda-Saavedra, D. Genome-wide enhancer prediction from epigenetic signatures using genetic algorithm-optimized support vector machines. Nucleic Acids Res. 2012, 40, e77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleftogiannis, D.; Kalnis, P.; Bajic, V.B. DEEP: A general computational framework for predicting enhancers. Nucleic Acids Res. 2015, 43, e6. [Google Scholar] [CrossRef] [Green Version]
- Firpi, H.A.; Ucar, D.; Tan, K. Discover regulatory DNA elements using chromatin signatures and artificial neural network. Bioinformatics 2010, 26, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Qu, W.; Shan, G.; Zhang, C. DELTA: A Distal Enhancer Locating Tool Based on AdaBoost Algorithm and Shape Features of Chromatin Modifications. PLoS ONE 2015, 10, e0130622. [Google Scholar] [CrossRef]
- Rajagopal, N.; Xie, W.; Li, Y.; Wagner, U.; Wang, W.; Stamatoyannopoulos, J.; Ernst, J.; Kellis, M.; Ren, B. RFECS: A random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput. Biol. 2013, 9, e1002968. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, M.M.; Buske, O.J.; Wang, J.; Weng, Z.; Bilmes, J.A.; Noble, W.S. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods 2012, 9, 473–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Fish, A.E.; Capra, J.A. Prediction of gene regulatory enhancers across species reveals evolutionarily conserved sequence properties. PLoS Comput. Biol. 2018, 14, e1006484. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Shi, W.; Wasserman, W.W. Genome-wide prediction of cis-regulatory regions using supervised deep learning methods. BMC Bioinform. 2018, 19, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Li, H.; Ren, C.; Bo, X.; Shu, W. PEDLA: Predicting enhancers with a deep learning-based algorithmic framework. Sci. Rep. 2016, 6, 28517. [Google Scholar] [CrossRef]
- Min, X.; Zeng, W.; Chen, S.; Chen, N.; Chen, T.; Jiang, R. Predicting enhancers with deep convolutional neural networks. BMC Bioinform. 2017, 18, 478. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Liu, F.; Ren, C.; Ouyang, Z.; Xie, Z.; Bo, X.; Shu, W. BiRen: Predicting enhancers with a deep-learning-based model using the DNA sequence alone. Bioinformatics 2017, 33, 1930–1936. [Google Scholar] [CrossRef] [Green Version]
- Encode Project Consortium; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Abugessaisa, I.; Ramilowski, J.A.; Lizio, M.; Severin, J.; Hasegawa, A.; Harshbarger, J.; Kondo, A.; Noguchi, S.; Yip, C.W.; Ooi, J.L.C.; et al. FANTOM enters 20th year: Expansion of transcriptomic atlases and functional annotation of non-coding RNAs. Nucleic Acids Res. 2021, 49, D892–D898. [Google Scholar] [CrossRef]
- Lee, D.; Karchin, R.; Beer, M.A. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011, 21, 2167–2180. [Google Scholar] [CrossRef] [Green Version]
- Ghandi, M.; Lee, D.; Mohammad-Noori, M.; Beer, M.A. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol. 2014, 10, e1003711. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Li, K.; Huang, D.S.; Chou, K.C. iEnhancer-EL: Identifying enhancers and their strength with ensemble learning approach. Bioinformatics 2018, 34, 3835–3842. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Nguyen-Vo, T.H.; Le, N.Q.K.; Do, T.T.T.; Rahardja, S.; Nguyen, B.P. iEnhancer-ECNN: Identifying enhancers and their strength using ensembles of convolutional neural networks. BMC Genom. 2019, 20, 951. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Yapp, E.K.Y.; Ho, Q.T.; Nagasundaram, N.; Ou, Y.Y.; Yeh, H.Y. iEnhancer-5Step: Identifying enhancers using hidden information of DNA sequences via Chou’s 5-step rule and word embedding. Anal. Biochem. 2019, 571, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Huangfu, D. Decoding the noncoding genome via large-scale CRISPR screens. Curr. Opin. Genet. Dev. 2018, 52, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, M.; Jayasurya, K.; Tomancak, P.; Ohler, U. An alignment-free method to identify candidate orthologous enhancers in multiple Drosophila genomes. Bioinformatics 2010, 26, 2109–2115. [Google Scholar] [CrossRef] [Green Version]
- Kazemian, M.; Suryamohan, K.; Chen, J.-Y.; Zhang, Y.; Samee, M.A.H.; Halfon, M.S.; Sinha, S. Evidence for Deep Regulatory Similarities in Early Developmental Programs across Highly Diverged Insects. Genome Biol. Evol. 2014, 6, 2301–2320. [Google Scholar] [CrossRef] [Green Version]
- Minnoye, L.; Taskiran, I.I.; Mauduit, D.; Fazio, M.; Van Aerschot, L.; Hulselmans, G.; Christiaens, V.; Makhzami, S.; Seltenhammer, M.; Karras, P.; et al. Cross-species analysis of enhancer logic using deep learning. Genome Res. 2020, 30, 1815–1834. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Bork, P. Quantification of insect genome divergence. Trends Genet. 2007, 23, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Cande, J.; Goltsev, Y.; Levine, M.S. Conservation of enhancer location in divergent insects. Proc. Natl. Acad. Sci. USA 2009, 106, 14414–14419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erives, A.; Levine, M. Coordinate enhancers share common organizational features in the Drosophila genome. Proc. Natl. Acad. Sci. USA 2004, 101, 3851–3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinzen, R.P.; Cande, J.; Ronshaugen, M.; Papatsenko, D.; Levine, M. Evolution of the ventral midline in insect embryos. Dev. Cell 2006, 11, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Goltsev, Y.; Fuse, N.; Frasch, M.; Zinzen, R.P.; Lanzaro, G.; Levine, M. Evolution of the dorsal-ventral patterning network in the mosquito, Anopheles gambiae. Development 2007, 134, 2415–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suryamohan, K.; Halfon, M.S. Overview Article: Identifying transcriptional cis-regulatory modules in animal genomes. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 59–84. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.T.; Deem, K.D.; Borras-Castells, F.; Sambrani, N.; Rudolf, H.; Suryamohan, K.; El-Sherif, E.; Halfon, M.S.; McKay, D.J.; Tomoyasu, Y. Enhancer identification and activity evaluation in the red flour beetle, Tribolium castaneum. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.; Reeve, S.; Grumbling, G.; Osumi-Sutherland, D. The Drosophila anatomy ontology. J. Biomed. Semant. 2013, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- De Renzis, S.; Elemento, O.; Tavazoie, S.; Wieschaus, E.F. Unmasking activation of the zygotic genome using chromosomal deletions in the Drosophila embryo. PLoS Biol. 2007, 5, e117. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhu, Q.; He, X.; Sinha, S.; Halfon, M.S. Large-scale analysis of transcriptional cis-regulatory modules reveals both common features and distinct subclasses. Genome Biol. 2007, 8, R101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papatsenko, D.; Goltsev, Y.; Levine, M. Organization of developmental enhancers in the Drosophila embryo. Nucleic Acids Res. 2009, 37, 5665–5677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinzen, R.P.; Girardot, C.; Gagneur, J.; Braun, M.; Furlong, E.E. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature 2009, 462, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Erceg, J.; Pakozdi, T.; Marco-Ferreres, R.; Ghavi-Helm, Y.; Girardot, C.; Bracken, A.P.; Furlong, E.E. Dual functionality of cis-regulatory elements as developmental enhancers and Polycomb response elements. Genes Dev. 2017, 31, 590–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blick, A.J.; Mayer-Hirshfeld, I.; Malibiran, B.R.; Cooper, M.A.; Martino, P.A.; Johnson, J.E.; Bateman, J.R. The Capacity to Act in Trans Varies among Drosophila Enhancers. Genetics 2016, 203, 203–218. [Google Scholar] [CrossRef] [Green Version]
- Vincent, B.J.; Staller, M.V.; Lopez-Rivera, F.; Bragdon, M.D.J.; Pym, E.C.G.; Biette, K.M.; Wunderlich, Z.; Harden, T.T.; Estrada, J.; DePace, A.H. Hunchback is counter-repressed to regulate even-skipped stripe 2 expression in Drosophila embryos. PLoS Genet. 2018, 14, e1007644. [Google Scholar] [CrossRef]
- Samee, M.A.H.; Lydiard-Martin, T.; Biette, K.M.; Vincent, B.J.; Bragdon, M.D.; Eckenrode, K.B.; Wunderlich, Z.; Estrada, J.; Sinha, S.; DePace, A.H. Quantitative Measurement and Thermodynamic Modeling of Fused Enhancers Support a Two-Tiered Mechanism for Interpreting Regulatory DNA. Cell Rep. 2017, 21, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Gisselbrecht, S.S.; Palagi, A.; Kurland, J.V.; Rogers, J.M.; Ozadam, H.; Zhan, Y.; Dekker, J.; Bulyk, M.L. Transcriptional Silencers in Drosophila Serve a Dual Role as Transcriptional Enhancers in Alternate Cellular Contexts. Mol. Cell 2020, 77, 324–337.e8. [Google Scholar] [CrossRef]
- Soluri, I.V.; Zumerling, L.M.; Payan Parra, O.A.; Clark, E.G.; Blythe, S.A. Zygotic pioneer factor activity of Odd-paired/Zic is necessary for late function of the Drosophila segmentation network. eLife 2020, 9. [Google Scholar] [CrossRef]
- Li, X.Y.; MacArthur, S.; Bourgon, R.; Nix, D.; Pollard, D.A.; Iyer, V.N.; Hechmer, A.; Simirenko, L.; Stapleton, M.; Luengo Hendriks, C.L.; et al. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 2008, 6, e27. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Y.; Thomas, S.; Sabo, P.J.; Eisen, M.B.; Stamatoyannopoulos, J.A.; Biggin, M.D. The role of chromatin accessibility in directing the widespread, overlapping patterns of Drosophila transcription factor binding. Genome Biol. 2011, 12, R34. [Google Scholar] [CrossRef] [Green Version]
- MacArthur, S.; Li, X.Y.; Li, J.; Brown, J.B.; Chu, H.C.; Zeng, L.; Grondona, B.P.; Hechmer, A.; Simirenko, L.; Keranen, S.V.; et al. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol. 2009, 10, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negre, N.; Brown, C.D.; Shah, P.K.; Kheradpour, P.; Morrison, C.A.; Henikoff, J.G.; Feng, X.; Ahmad, K.; Russell, S.; White, R.A.; et al. A comprehensive map of insulator elements for the Drosophila genome. PLoS Genet. 2010, 6, e1000814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moshkovich, N.; Nisha, P.; Boyle, P.J.; Thompson, B.A.; Dale, R.K.; Lei, E.P. RNAi-independent role for Argonaute2 in CTCF/CP190 chromatin insulator function. Genes Dev. 2011, 25, 1686–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoroshko, V.A.; Levitsky, V.G.; Zykova, T.Y.; Antonenko, O.V.; Belyaeva, E.S.; Zhimulev, I.F. Chromatin Heterogeneity and Distribution of Regulatory Elements in the Late-Replicating Intercalary Heterochromatin Domains of Drosophila melanogaster Chromosomes. PLoS ONE 2016, 11, e0157147. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Probabilistic modelling of chromatin code landscape reveals functional diversity of enhancer-like chromatin states. Nat. Commun. 2016, 7, 10528. [Google Scholar] [CrossRef] [Green Version]
- Mateo, L.J.; Murphy, S.E.; Hafner, A.; Cinquini, I.S.; Walker, C.A.; Boettiger, A.N. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 2019, 568, 49–54. [Google Scholar] [CrossRef]
- Bozek, M.; Cortini, R.; Storti, A.E.; Unnerstall, U.; Gaul, U.; Gompel, N. ATAC-seq reveals regional differences in enhancer accessibility during the establishment of spatial coordinates in the Drosophila blastoderm. Genome Res. 2019, 29, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Ghavi-Helm, Y.; Klein, F.A.; Pakozdi, T.; Ciglar, L.; Noordermeer, D.; Huber, W.; Furlong, E.E. Enhancer loops appear stable during development and are associated with paused polymerase. Nature 2014, 512, 96–100. [Google Scholar] [CrossRef]
- Li, L.; Wunderlich, Z. An Enhancer’s Length and Composition Are Shaped by Its Regulatory Task. Front. Genet. 2017, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Schor, I.E.; Bussotti, G.; Males, M.; Forneris, M.; Viales, R.R.; Enright, A.J.; Furlong, E.E.M. Non-coding RNA Expression, Function, and Variation during Drosophila Embryogenesis. Curr. Biol. 2018, 28, 3547–3561.e9. [Google Scholar] [CrossRef]
- Mikhaylichenko, O.; Bondarenko, V.; Harnett, D.; Schor, I.E.; Males, M.; Viales, R.R.; Furlong, E.E.M. The degree of enhancer or promoter activity is reflected by the levels and directionality of eRNA transcription. Genes Dev. 2018, 32, 42–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haines, J.E.; Eisen, M.B. Patterns of chromatin accessibility along the anterior-posterior axis in the early Drosophila embryo. PLoS Genet. 2018, 14, e1007367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusanovich, D.A.; Reddington, J.P.; Garfield, D.A.; Daza, R.M.; Aghamirzaie, D.; Marco-Ferreres, R.; Pliner, H.A.; Christiansen, L.; Qiu, X.; Steemers, F.J.; et al. The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 2018, 555, 538–542. [Google Scholar] [CrossRef]
- Costello, J.C.; Dalkilic, M.M.; Beason, S.M.; Gehlhausen, J.R.; Patwardhan, R.; Middha, S.; Eads, B.D.; Andrews, J.R. Gene networks in Drosophila melanogaster: Integrating experimental data to predict gene function. Genome Biol. 2009, 10, R97. [Google Scholar] [CrossRef] [Green Version]
- Kazemian, M.; Blatti, C.; Richards, A.; McCutchan, M.; Wakabayashi-Ito, N.; Hammonds, A.S.; Celniker, S.E.; Kumar, S.; Wolfe, S.A.; Brodsky, M.H.; et al. Quantitative analysis of the Drosophila segmentation regulatory network using pattern generating potentials. PLoS Biol. 2010, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marbach, D.; Roy, S.; Ay, F.; Meyer, P.E.; Candeias, R.; Kahveci, T.; Bristow, C.A.; Kellis, M. Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Res. 2012, 22, 1334–1349. [Google Scholar] [CrossRef] [Green Version]
- Pesch, R.; Zimmer, R. Cross-species Conservation of context-specific networks. BMC Syst. Biol. 2016, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Reda, C.; Wilczynski, B. Automated inference of gene regulatory networks using explicit regulatory modules. J. Theor. Biol. 2020, 486, 110091. [Google Scholar] [CrossRef]
- Yang, B.; Wittkopp, P.J. Structure of the Transcriptional Regulatory Network Correlates with Regulatory Divergence in Drosophila. Mol. Biol. Evol. 2017, 34, 1352–1362. [Google Scholar] [CrossRef] [Green Version]
- Drosophila 12 Genomes Consortium; Clark, A.G.; Eisen, M.B.; Smith, D.R.; Bergman, C.M.; Oliver, B.; Markow, T.A.; Kaufman, T.C.; Kellis, M.; Gelbart, W.; et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature 2007, 450, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Hare, E.E.; Peterson, B.K.; Iyer, V.N.; Meier, R.; Eisen, M.B. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 2008, 4, e1000106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.Z.; Holloway, A.K.; Maerkl, S.J.; Kreitman, M. Does positive selection drive transcription factor binding site turnover? A test with Drosophila cis-regulatory modules. PLoS Genet. 2011, 7, e1002053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, A.K.; Begun, D.J.; Siepel, A.; Pollard, K.S. Accelerated sequence divergence of conserved genomic elements in Drosophila melanogaster. Genome Res. 2008, 18, 1592–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Ludwig, M.Z.; Kreitman, M.; Reinitz, J. Natural variation of the expression pattern of the segmentation gene even-skipped in melanogaster. Dev. Biol. 2015, 405, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Khoueiry, P.; Girardot, C.; Ciglar, L.; Peng, P.C.; Gustafson, E.H.; Sinha, S.; Furlong, E.E. Uncoupling evolutionary changes in DNA sequence, transcription factor occupancy and enhancer activity. eLife 2017, 6. [Google Scholar] [CrossRef]
- Macdonald, S.J.; Long, A.D. Fine scale structural variants distinguish the genomes of Drosophila melanogaster and D. pseudoobscura. Genome Biol. 2006, 7, R67. [Google Scholar] [CrossRef] [Green Version]
- Aerts, S.; van Helden, J.; Sand, O.; Hassan, B.A. Fine-tuning enhancer models to predict transcriptional targets across multiple genomes. PLoS ONE 2007, 2, e1115. [Google Scholar] [CrossRef]
- Brody, T.; Rasband, W.; Baler, K.; Kuzin, A.; Kundu, M.; Odenwald, W.F. cis-Decoder discovers constellations of conserved DNA sequences shared among tissue-specific enhancers. Genome Biol. 2007, 8, R75. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Huo, H.; Yu, Q. SMCis: An Effective Algorithm for Discovery of Cis-Regulatory Modules. PLoS ONE 2016, 11, e0162968. [Google Scholar] [CrossRef] [PubMed]
- Ivan, A.; Halfon, M.S.; Sinha, S. Computational discovery of cis-regulatory modules in Drosophila without prior knowledge of motifs. Genome Biol. 2008, 9, R22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Teichmann, S.A.; Down, T.A. Assessing computational methods of cis-regulatory module prediction. PLoS Comput. Biol. 2010, 6, e1001020. [Google Scholar] [CrossRef] [PubMed]
- Arbel, H.; Basu, S.; Fisher, W.W.; Hammonds, A.S.; Wan, K.H.; Park, S.; Weiszmann, R.; Booth, B.W.; Keranen, S.V.; Henriquez, C.; et al. Exploiting regulatory heterogeneity to systematically identify enhancers with high accuracy. Proc. Natl. Acad. Sci. USA 2019, 116, 900–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schember, I.; Halfon, M.S. Identification of new Anopheles gambiae transcriptional enhancers using a cross-species prediction approach. Insect Mol. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- NCBI Assembly. Available online: https://www.ncbi.nlm.nih.gov/assembly/GCA_001676475.1 (accessed on 26 May 2021).
- i5K Sequenced Arthropod Genomes. Available online: http://i5k.github.io/arthropod_genomes_at_ncbi (accessed on 26 May 2021).
- Hotaling, S.; Sproul, J.S.; Heckenhauer, J.; Powell, A.; Larracuente, A.M.; Pauls, S.U.; Kelley, J.L.; Frandsen, P.B. Long-reads are revolutionizing 20 years of insect genome sequencing. Genome Biol. Evol. 2021. [Google Scholar] [CrossRef]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, Z.; Bragdon, M.D.; Vincent, B.J.; White, J.A.; Estrada, J.; DePace, A.H. Kruppel Expression Levels Are Maintained through Compensatory Evolution of Shadow Enhancers. Cell Rep. 2015, 12, 1740–1747. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, E.; Khoueiry, P.; Garfield, D.A.; Geeleher, P.; Zichner, T.; Gustafson, E.H.; Ciglar, L.; Korbel, J.O.; Furlong, E.E. Shadow Enhancers Are Pervasive Features of Developmental Regulatory Networks. Curr. Biol. 2016, 26, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gontarz, P.; Fu, S.; Xing, X.; Liu, S.; Miao, B.; Bazylianska, V.; Sharma, A.; Madden, P.; Cates, K.; Yoo, A.; et al. Comparison of differential accessibility analysis strategies for ATAC-seq data. Sci. Rep. 2020, 10, 10150. [Google Scholar] [CrossRef]
- Martins, A.L.; Walavalkar, N.M.; Anderson, W.D.; Zang, C.; Guertin, M.J. Universal correction of enzymatic sequence bias reveals molecular signatures of protein/DNA interactions. Nucleic Acids Res. 2018, 46, e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orchard, P.; Kyono, Y.; Hensley, J.; Kitzman, J.O.; Parker, S.C.J. Quantification, Dynamic Visualization, and Validation of Bias in ATAC-Seq Data with ataqv. Cell Syst. 2020, 10, 298–306.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.R.; Quach, B.; Furey, T.S. Correcting nucleotide-specific biases in high-throughput sequencing data. BMC Bioinform. 2017, 18, 357. [Google Scholar] [CrossRef]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asma, H.; Halfon, M.S. Annotating the Insect Regulatory Genome. Insects 2021, 12, 591. https://doi.org/10.3390/insects12070591
Asma H, Halfon MS. Annotating the Insect Regulatory Genome. Insects. 2021; 12(7):591. https://doi.org/10.3390/insects12070591
Chicago/Turabian StyleAsma, Hasiba, and Marc S. Halfon. 2021. "Annotating the Insect Regulatory Genome" Insects 12, no. 7: 591. https://doi.org/10.3390/insects12070591
APA StyleAsma, H., & Halfon, M. S. (2021). Annotating the Insect Regulatory Genome. Insects, 12(7), 591. https://doi.org/10.3390/insects12070591