
Transcriptome Analysis of Hormone-and Cuticle-Related Genes in the Development Process of Deutonymph in Tetranychus urticae


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mite Culture
2.2. RNA Isolation, and Transcriptome Sequencing
2.3. Analysis of RNA-Seq Data
2.4. Differential Expression Analysis
2.5. Expression Dynamics Analysis of Differential Expression Genes in Development Process
3. Results
3.1. RNA-Seq Data
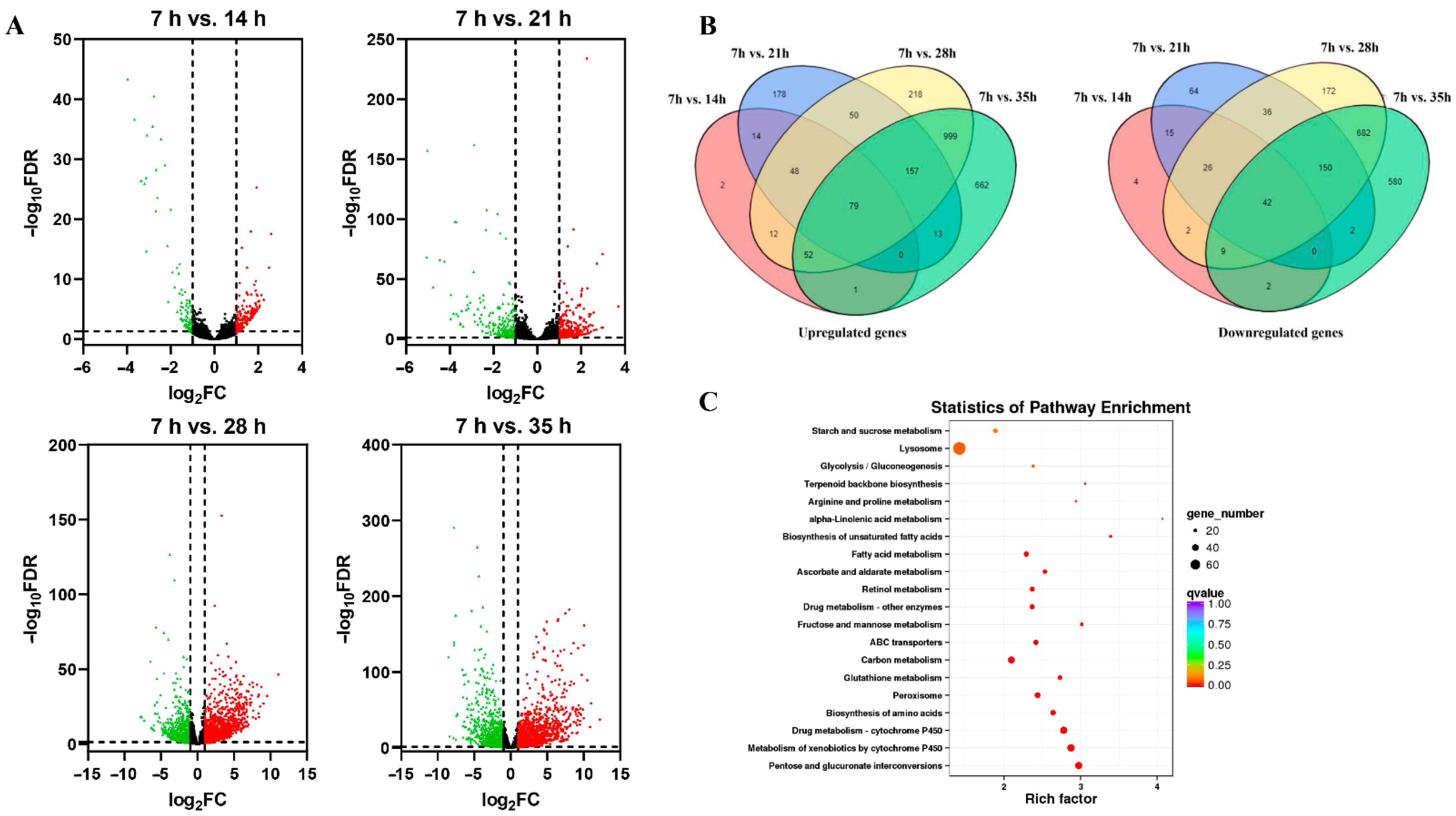
3.2. Differential Expression Genes Analysis in Development Process of Deutonymph in T. urticae
3.3. Function Analysis of Differential Expression Genes in Development Process of Deutonymph
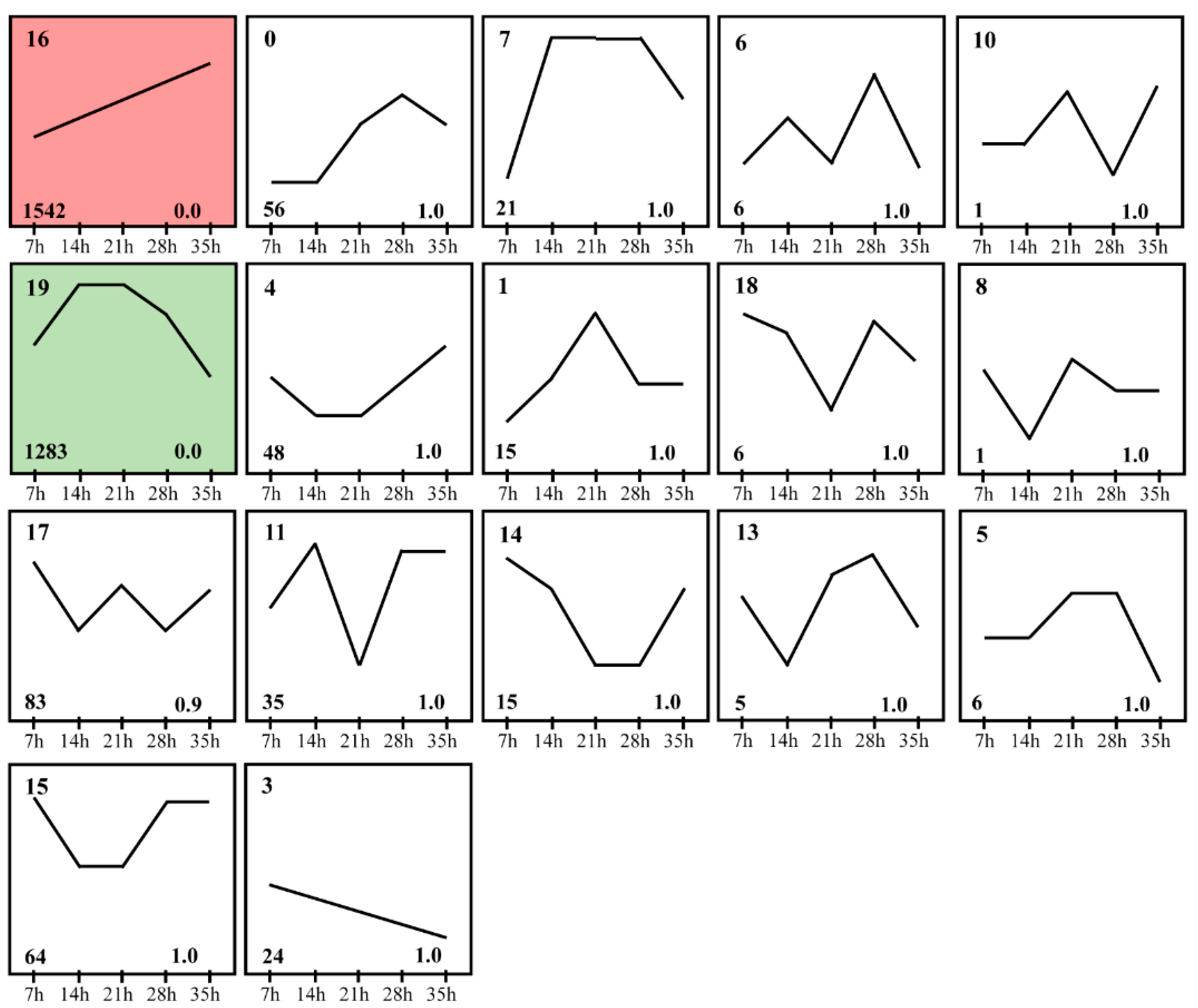
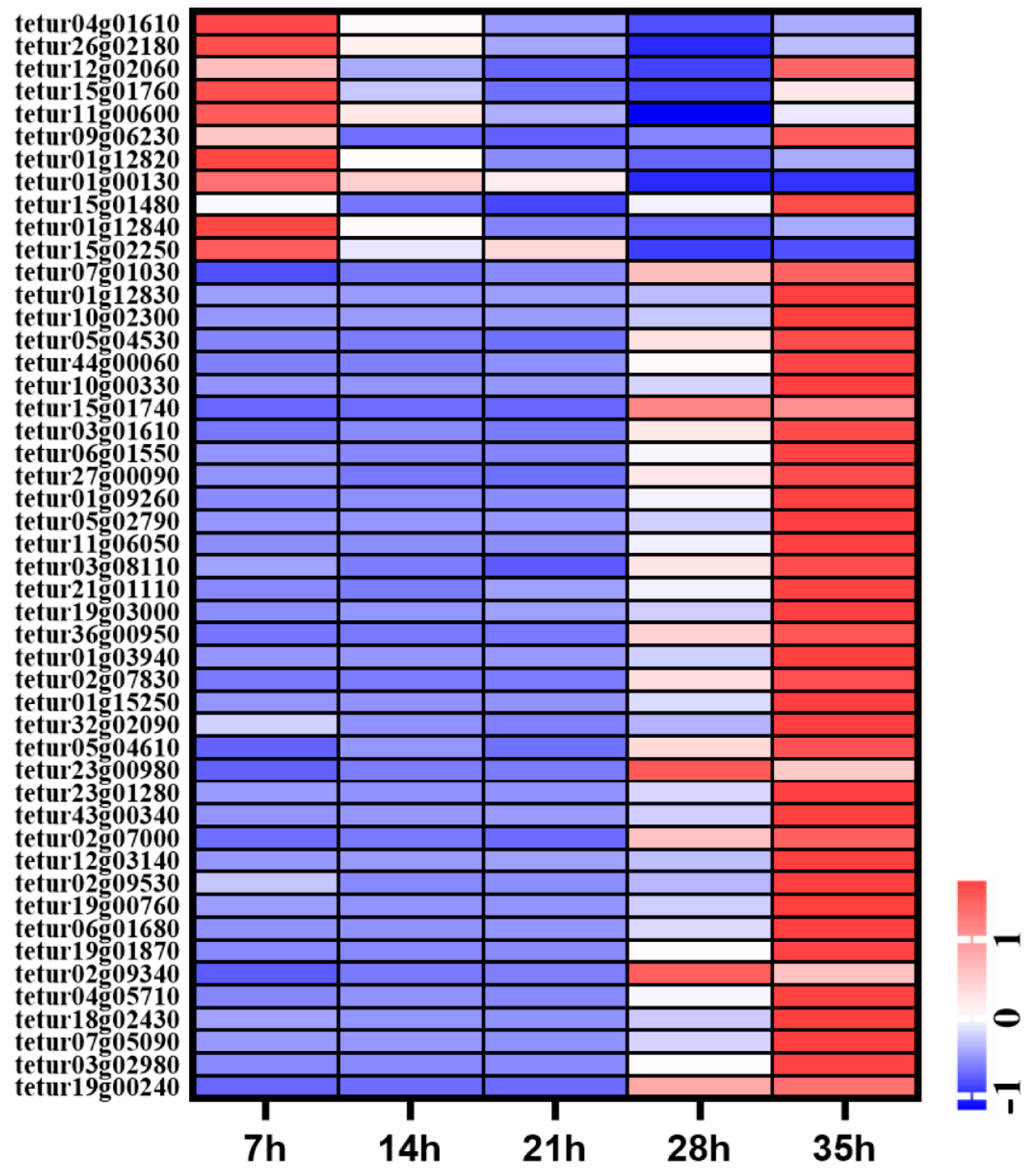
3.4. Expression Patters of mRNAs in Development Process of Deutonymph
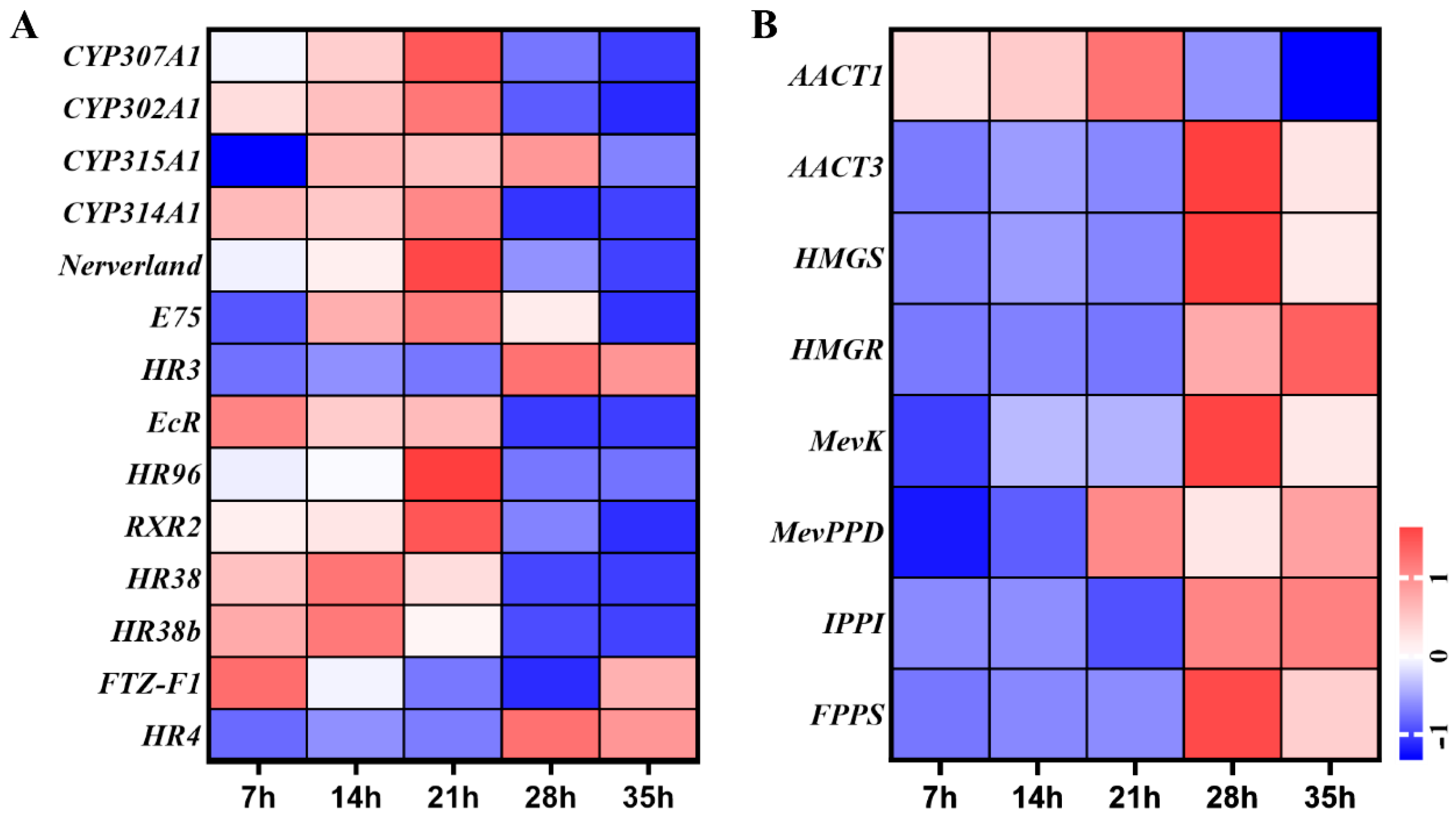
3.5. Expression Level Changes of Ecdysteroid and Juvenile Hormones Pathways Genes
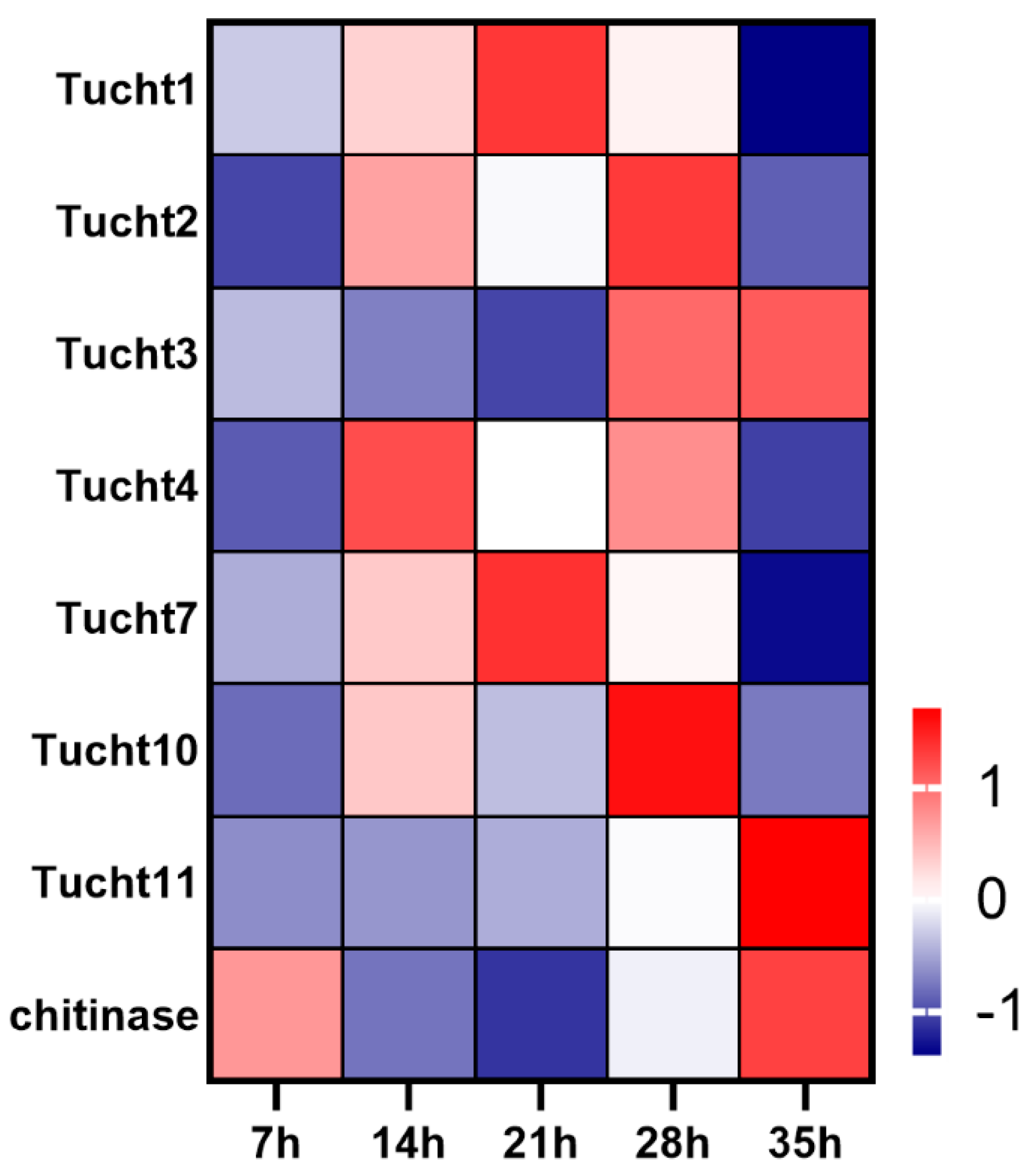
3.6. Expression Dynamics of Chitin Related Pathways Genes during Development Process of Deutonymph
3.7. Expression Pattern of Cuticle Protein Genes in Different Development Time Points
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamanaka, N.; Rewitz, K.F.; O’Connor, M.B. Ecdysone control of developmental transitions: Lessons from Drosophila research. Annu. Rev. Entomol. 2013, 58, 497–516. [Google Scholar] [CrossRef] [Green Version]
- Moussian, B. Recent advances in understanding mechanisms of insect cuticle differentiation. Insect Biochem. Mol. Biol. 2010, 40, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Omar, M.A.A.; Ao, Y.; Li, M.; He, K.; Xu, L.; Tong, H.; Jiang, M.; Li, F. The functional difference of eight chitinase genes between male and female of the cotton mealybug, Phenacoccus solenopsis. Insect Mol. Biol. 2019, 28, 550–567. [Google Scholar] [CrossRef]
- Kramer, K.J.; Muthukrishnan, S. Insect chitinases: Molecular biology and potential use as biopesticides. Insect Biochem. Mol. Biol. 1997, 27, 887–900. [Google Scholar] [CrossRef]
- Ostrowski, S.; Dierick, H.A.; Bejsovec, A. Genetic control of cuticle formation during embryonic development of Drosophila melanogaster. Genetics 2002, 161, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Yang, Q. Physiological significance of alternatively spliced exon combinations of the single-copy gene class A chitin synthase in the insect Ostrinia furnacalis (Lepidoptera). Insect Mol. Biol. 2012, 21, 395–404. [Google Scholar] [CrossRef]
- Cohen, E. Chitin synthesis and inhibition: A revisit. Pest Manag. Sci. 2001, 57, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Doucet, D.; Retnakaran, A. Insect chitin: Metabolism, genomics and pest management. Adv. Insect Physiol. 2012, 43, 437–511. [Google Scholar]
- Huang, Q.S.; Xie, X.L.; Liang, G.; Gong, F.; Wang, Y.; Wei, X.Q.; Wang, Q.; Ji, Z.L.; Chen, Q.X. The GH18 family of chitinases: Their domain architectures, functions and evolutions. Glycobiology 2012, 22, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Rebers, J.E.; Willis, J.H. A conserved domain in arthropod cuticular proteins binds chitin. Insect Biochem. Mol. Biol. 2001, 31, 1083–1093. [Google Scholar] [CrossRef]
- Andersen, S.O. Studies on proteins in post-ecdysial nymphal cuticle of locust, Locusta migratoria, and cockroach, Blaberus craniifer. Insect Biochem. Mol. Biol. 2000, 30, 569–577. [Google Scholar] [CrossRef]
- Zhou, Y.; Badgett, M.J.; Bowen, J.H.; Vannini, L.; Orlando, R.; Willis, J.H. Distribution of cuticular proteins in different structures of adult Anopheles gambiae. Insect Biochem. Mol. Biol. 2016, 75, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, R.; Tanaka, S.; Shiotsuki, T. RNAi-mediated knockdown of SPOOK reduces ecdysteroid titers and causes precocious metamorphosis in the desert locust Schistocerca gregaria. Dev. Biol. 2017, 429, 71–80. [Google Scholar] [CrossRef]
- Li, G.; Sun, Q.Z.; Liu, X.Y.; Zhang, J.; Dou, W.; Niu, J.Z.; Wang, J.J. Expression dynamics of key ecdysteroid and juvenile hormone biosynthesis genes imply a coordinated regulation pattern in the molting process of a spider mite, Tetranychus urticae. Exp. Appl. Acarol. 2019, 78, 361–372. [Google Scholar] [CrossRef]
- Ali, M.W.; Khan, M.M.; Song, F.; Wu, L.; He, L.; Wang, Z.; Zhang, Z.Y.; Zhang, H.; Jiang, Y. RNA interference-based silencing of the Chitin Synthase 1 gene for reproductive and developmental disruptions in Panonychus citri. Insects 2020, 11, 786. [Google Scholar] [CrossRef]
- Xia, W.K.; Shen, X.M.; Ding, T.B.; Niu, J.Z.; Zhong, R.; Liao, C.Y.; Feng, Y.C.; Dou, W.; Wang, J.J. Functional analysis of a chitinase gene during the larval-nymph transition in Panonychus citri by RNA interference. Exp. Appl. Acarol. 2016, 70, 1–15. [Google Scholar] [CrossRef]
- Grbic, M.; Van Leeuwen, T.; Clark, R.M.; Rombauts, S.; Rouze, P.; Grbic, V.; Osborne, E.J.; Dermauw, W.; Ngoc, P.C.; Ortego, F.; et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature 2011, 479, 487–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, D.; Wang, F.; Xie, S.; Sun, F.; Wang, Z.; Peng, M.; Li, J. Developmental transcriptome analysis and identification of genes involved in larval metamorphosis of the razor clam, Sinonovacula constricta. Mar. Biotechnol. 2016, 18, 168–175. [Google Scholar] [CrossRef]
- Rahmat, N.L.; Zifruddin, A.N.; Zainal Abidin, C.M.R.; Nor Muhammad, N.A.; Hassan, M. The developmental transcriptome of bagworm, Metisa plana (Lepidoptera: Psychidae) and insights into chitin biosynthesis genes. Genes 2020, 12, 7. [Google Scholar] [CrossRef]
- Liu, S.H.; Xia, Y.D.; Zhang, Q.; Li, W.; Li, R.Y.; Liu, Y.; Chen, E.H.; Dou, W.; Stelinski, L.L.; Wang, J.J. Potential targets for controlling Bactrocera dorsalis using cuticle- and hormone-related genes revealed by a developmental transcriptome analysis. Pest Manag. Sci. 2020, 76, 2127–2143. [Google Scholar] [CrossRef]
- Chen, E.H.; Hou, Q.L.; Dou, W.; Wei, D.D.; Yue, Y.; Yang, R.L.; Yu, S.F.; De Schutter, K.; Smagghe, G.; Wang, J.J. RNA-seq analysis of gene expression changes during pupariation in Bactrocera dorsalis (Hendel) (Diptera: Tephritidae). BMC Genom. 2018, 19, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Guo, L.; Jiao, X.; Yang, N.; Yang, X.; Wu, Q.; Wang, S.; Zhou, X.; Zhang, Y. Transcriptomic dissection of sexual differences in Bemisia tabaci, an invasive agricultural pest worldwide. Sci. Rep. 2014, 4, 4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Li, K.; Gao, Y.; Liu, X.; Chen, W.; Ge, W.; Feng, Q.; Palli, S.R.; Li, S. Antagonistic actions of juvenile hormone and 20-hydroxyecdysone within the ring gland determine developmental transitions in Drosophila. Proc. Natl. Acad. Sci. USA 2018, 115, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Liu, X.Y.; Han, X.; Niu, J.Z.; Wang, J.J. RNAi of the nuclear receptor HR3 suggests a role in the molting process of the spider mite Panonychus citri. Exp. Appl. Acarol. 2020, 81, 75–83. [Google Scholar] [CrossRef]
- Kramer, K.J.; Corpuz, L.; Choi, H.K.; Muthukrishnan, S. Sequence of a cDNA and expression of the gene encoding epidermal and gut chitinases of Manduca sexta. Insect Biochem. Mol. Biol. 1993, 23, 691–701. [Google Scholar] [CrossRef]
- Zhao, X.; Qin, Z.; Liu, W.; Liu, X.; Moussian, B.; Ma, E.; Li, S.; Zhang, J. Nuclear receptor HR3 controls locust molt by regulating chitin synthesis and degradation genes of Locusta migratoria. Insect Biochem. Mol. Biol. 2017, 92, 1–11. [Google Scholar] [CrossRef]
- Wybouw, N.; Van Leeuwen, T.; Dermauw, W. A massive incorporation of microbial genes into the genome of Tetranychus urticae, a polyphagous arthropod herbivore. Insect Mol. Biol. 2018, 27, 333–351. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Replicates | Read Sum | Base Sum | GC (%) | Q30 (%) |
|---|---|---|---|---|---|
| 7 h | 1 | 19,966,666 | 5,964,221,328 | 39.22 | 93.38 |
| 2 | 23,132,933 | 6,910,020,364 | 39.05 | 93.19 | |
| 3 | 19,239,246 | 5,753,690,284 | 39.16 | 93.16 | |
| 14 h | 1 | 21,402,946 | 6,397,745,756 | 38.88 | 92.81 |
| 2 | 19,627,145 | 5,857,497,150 | 38.95 | 92.74 | |
| 3 | 21,981,138 | 6,588,408,854 | 37.49 | 92.34 | |
| 21 h | 1 | 23,280,649 | 6,901,943,666 | 38.76 | 92.51 |
| 2 | 20,689,137 | 6,184,409,592 | 38.97 | 93.47 | |
| 3 | 19,498,291 | 5,808,116,624 | 38.88 | 92.93 | |
| 28 h | 1 | 20,130,351 | 6,010,236,656 | 38.96 | 92.86 |
| 2 | 19,739,950 | 5,886,058,026 | 38.40 | 92.37 | |
| 3 | 19,485,760 | 5,825,531,426 | 38.31 | 92.72 | |
| 35 h | 1 | 20,756,485 | 6,203,250,222 | 39.17 | 93.26 |
| 2 | 20,604,866 | 6,166,389,390 | 38.62 | 92.48 | |
| 3 | 20,504,881 | 6,130,799,544 | 38.43 | 93.16 |
| DEG Set | Total | Annotated | GO | KEGG | COG | NR | eggNOG | Swiss-Prot | KOG | Pfam |
|---|---|---|---|---|---|---|---|---|---|---|
| 7 h vs. 14 h | 242 | 213 | 442 | 56 | 64 | 211 | 130 | 91 | 92 | 128 |
| 7 h vs. 21 h | 657 | 573 | 1270 | 267 | 237 | 569 | 397 | 298 | 342 | 380 |
| 7 h vs. 28 h | 2169 | 1855 | 1005 | 835 | 732 | 1845 | 1332 | 992 | 1191 | 1279 |
| 7 h vs. 35 h | 3075 | 2576 | 1421 | 1330 | 1030 | 2563 | 1871 | 1385 | 1676 | 1824 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Gu, X.; Gui, S.; Guo, J.; Yi, T.; Jin, D. Transcriptome Analysis of Hormone-and Cuticle-Related Genes in the Development Process of Deutonymph in Tetranychus urticae. Insects 2021, 12, 736. https://doi.org/10.3390/insects12080736
Li G, Gu X, Gui S, Guo J, Yi T, Jin D. Transcriptome Analysis of Hormone-and Cuticle-Related Genes in the Development Process of Deutonymph in Tetranychus urticae. Insects. 2021; 12(8):736. https://doi.org/10.3390/insects12080736
Chicago/Turabian StyleLi, Gang, Xinyao Gu, Shunhua Gui, Jianjun Guo, Tianci Yi, and Daochao Jin. 2021. "Transcriptome Analysis of Hormone-and Cuticle-Related Genes in the Development Process of Deutonymph in Tetranychus urticae" Insects 12, no. 8: 736. https://doi.org/10.3390/insects12080736
APA StyleLi, G., Gu, X., Gui, S., Guo, J., Yi, T., & Jin, D. (2021). Transcriptome Analysis of Hormone-and Cuticle-Related Genes in the Development Process of Deutonymph in Tetranychus urticae. Insects, 12(8), 736. https://doi.org/10.3390/insects12080736