On Oxidation Resistance Mechanisms at 1273 K of Tungsten-Based Alloys Containing Chromium and Yttria

 , ,
, ,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
- An overview of the employed experimental methods and the preparation of the alloys is given.
- The prepared alloys are pre-characterized.
- The changes during oxidation are analyzed, first on a macroscopic scale by measuring the the mass change, then on a microscopic scale by imaging the microstructure. This analysis investigates the protective oxide layer, discusses the main failure mechanism of the oxide, and predicts a lifetime.
- Further details on the microscopic properties are investigated: the diffusion processes yielding the formation of the protective oxide are analyzed and the phase formation, including its consequences, is studied.
- The distribution of alloying elements is analyzed on a nano-scale. A particular focus is on Y which is not detected in measurements on a microscopic scale.
2. Experimental Methods
2.1. Analysis Techniques
2.2. Sample Preparation
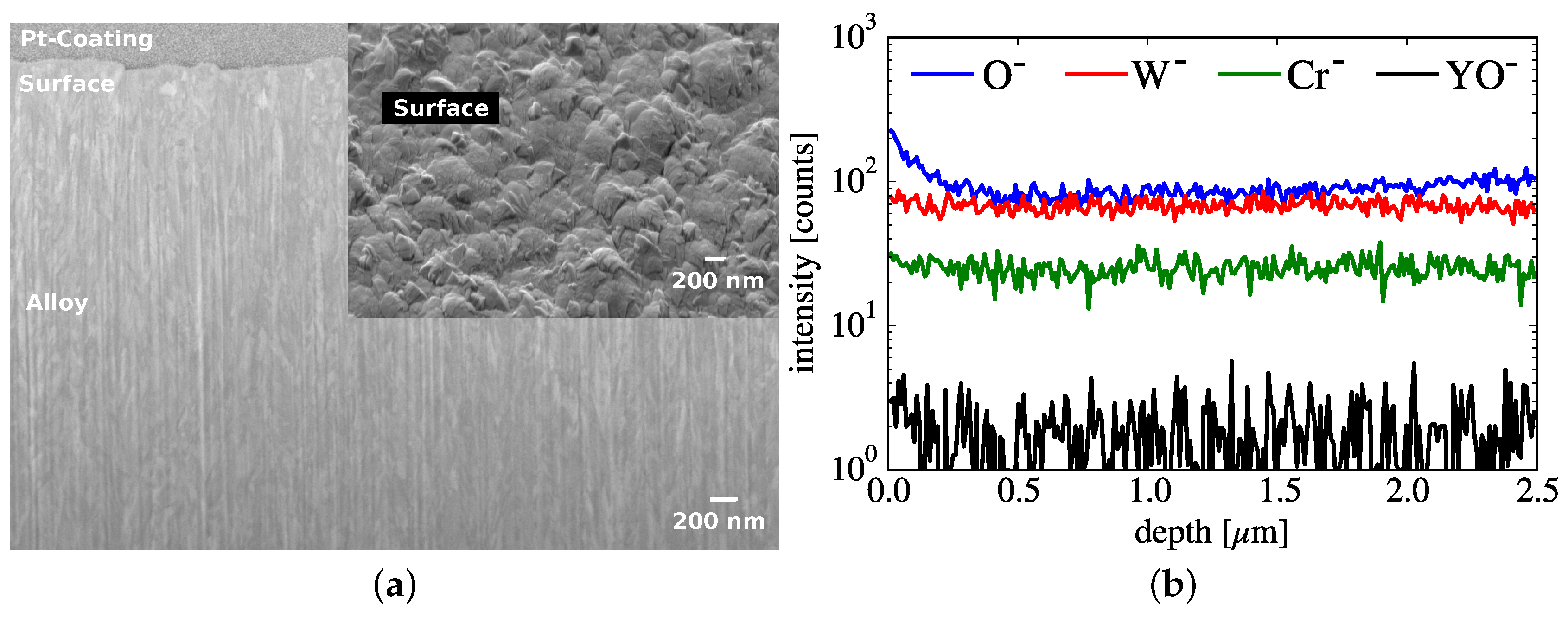
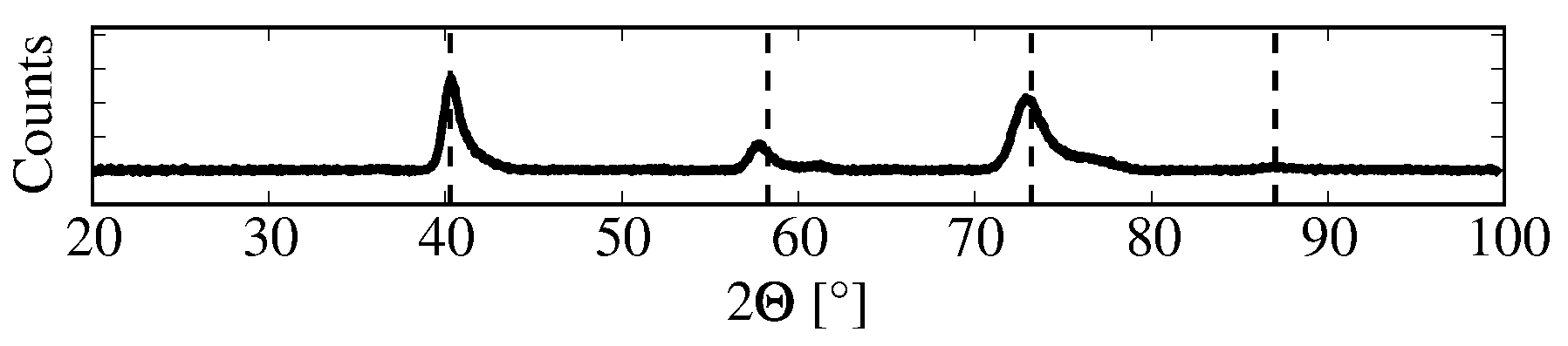
3. Sample Pre-Characterization
4. Macroscopic and Microscopic Changes During Oxidation
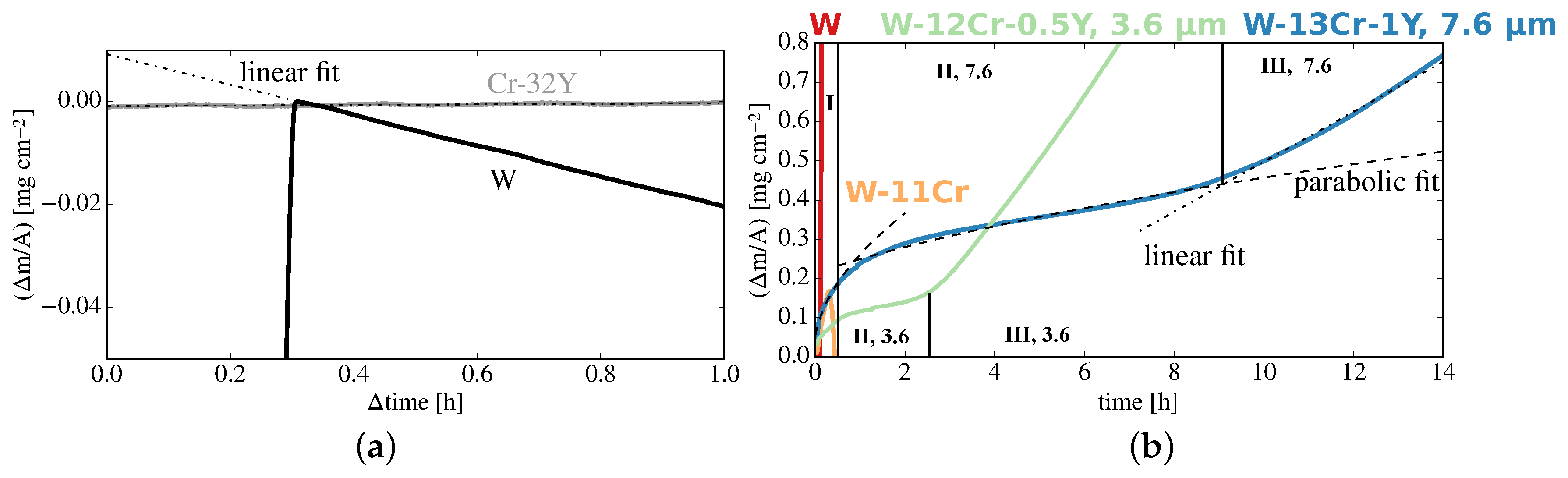
4.1. Macroscopic Changes: Mass Change
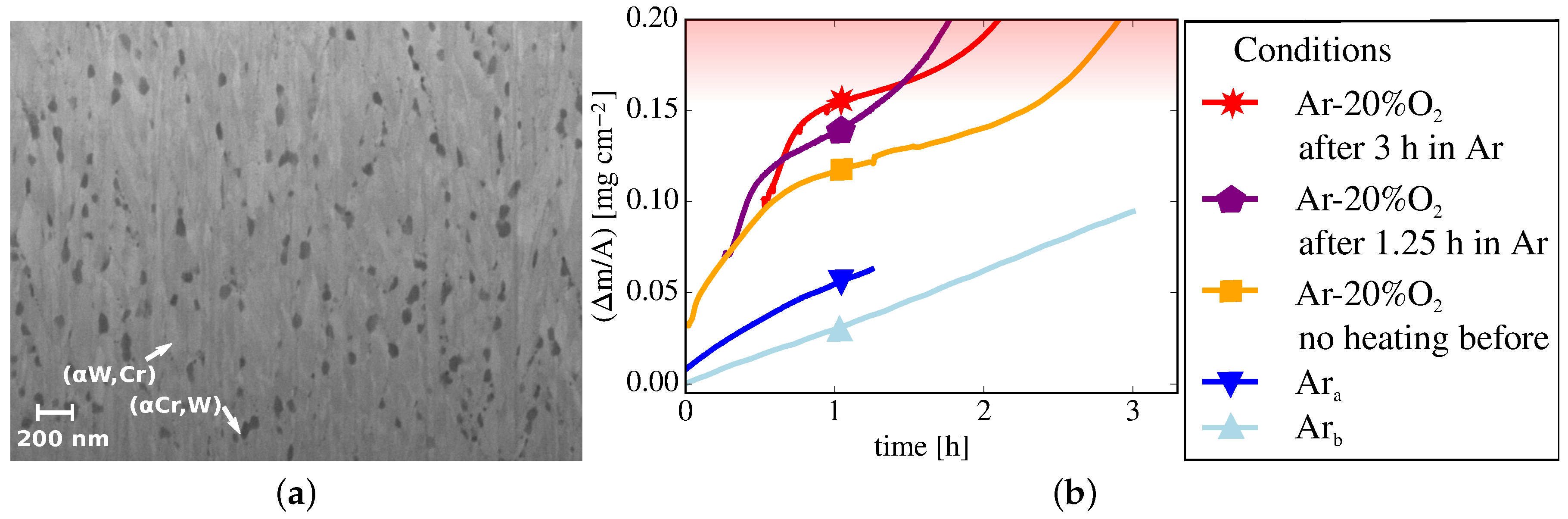
- Stage I lasts , the thick sample exhibits a parabolic oxidation rate of . In comparison, the rate for the thick sample with the optimized composition is 4 times lower.
- Stage II exhibits a parabolic oxidation rate reduced to for the thick sample. A gradient as low as is reached. Stage II ends after ; for a thinner sample, with a thickness of stage II lasted only under identical conditions despite the optimized composition and a lower oxidation rate of .
- Stage III follows for samples of either thickness. An increased mass gain with a linear rate of or is measured for the and thick samples, respectively.
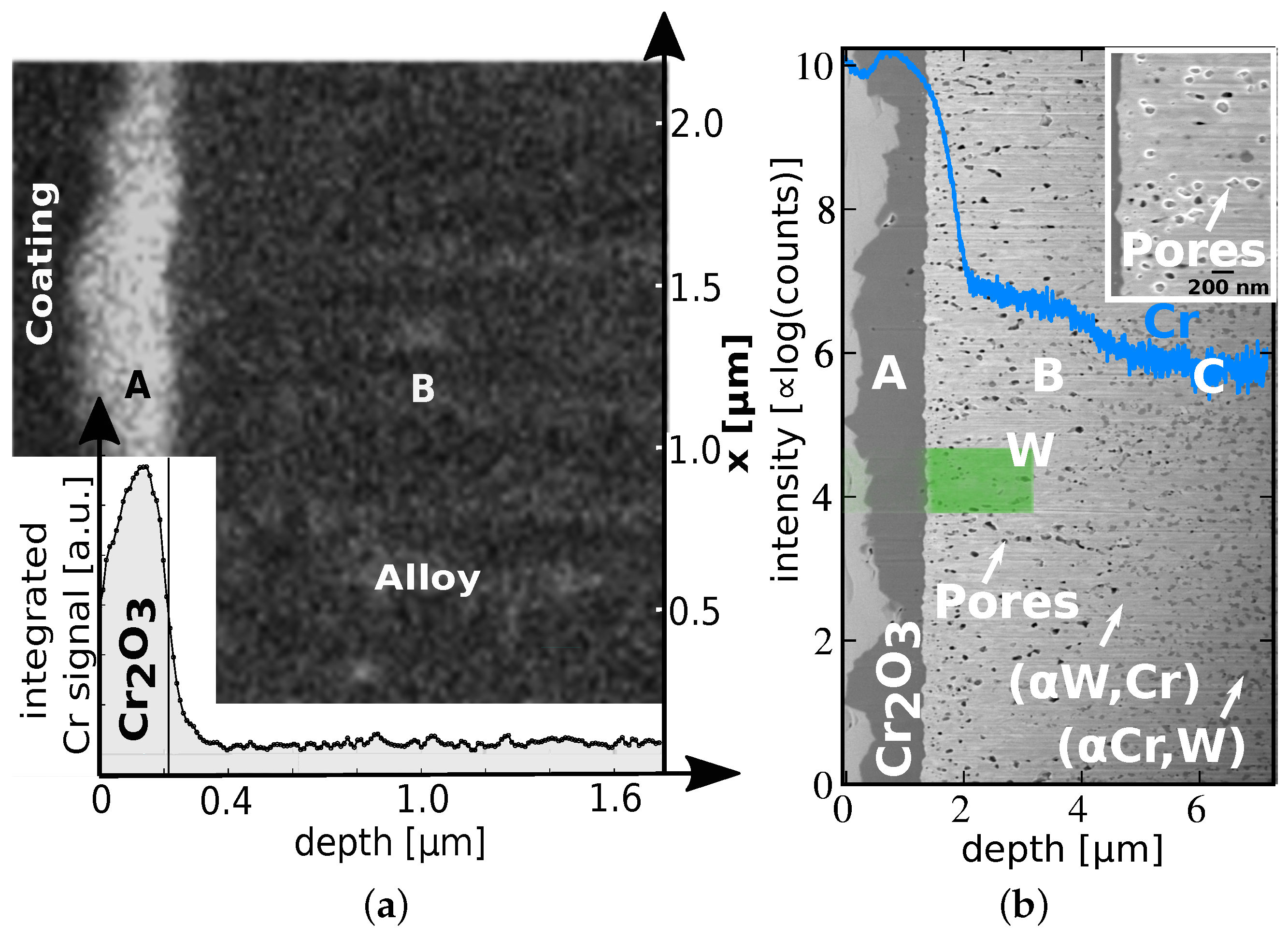
4.2. Microscopic Changes: Microstructure
4.3. Evaluation of the Oxidation Performance
- Is oxidation of tungsten suppressed?
- Does the chromium oxide sublimate/evaporate?
- How long can the material last?
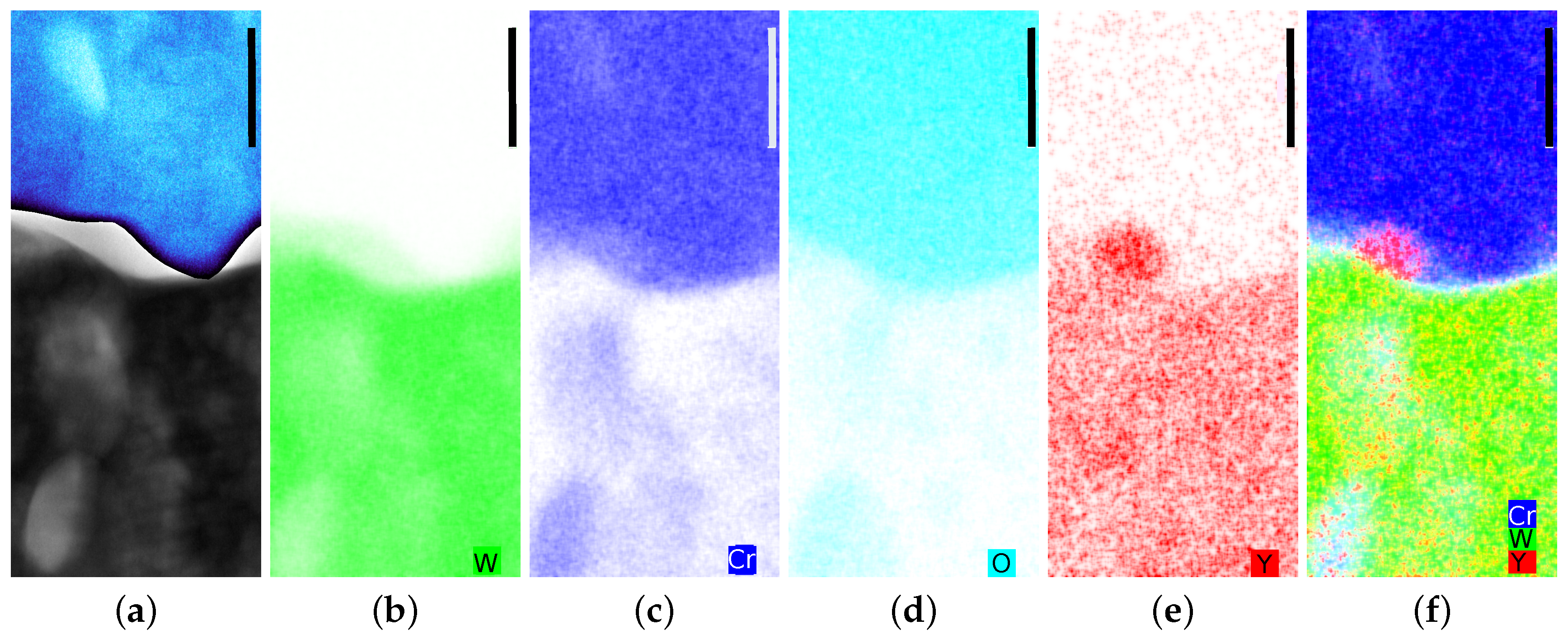
5. Microscopic Properties
5.1. On the Growth Direction of the Oxide
5.2. On Diffusion of Cr and Phase Formation
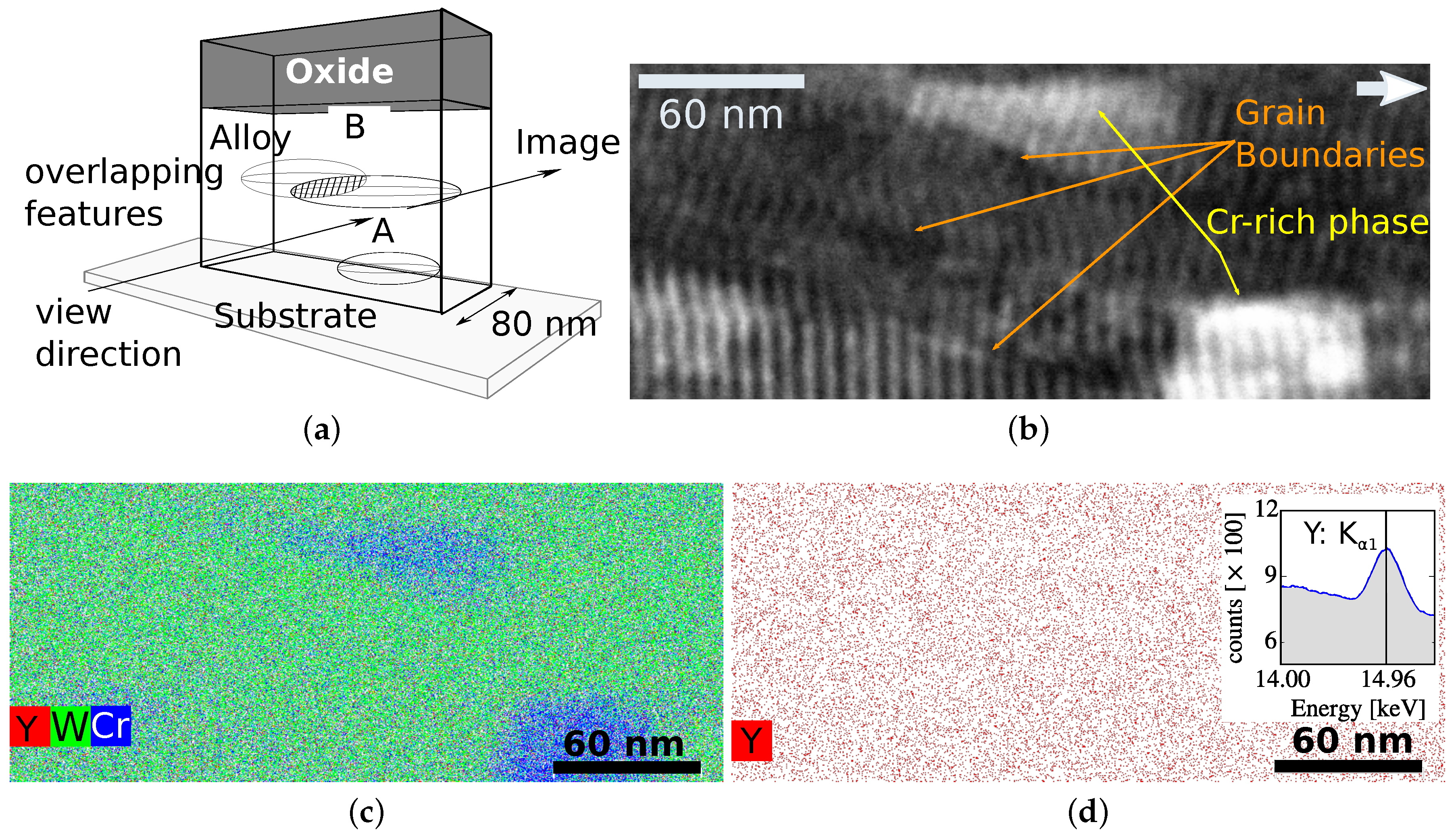
6. Investigations on a Nano-Scale
6.1. Results of a Scanning Transmission Electron Microscopy Study
6.2. Importance of Nano-Scale Effects for Understanding the Oxidation Resistance
- The nucleation sites are crucial for the improved oxidation resistance, as discussed above.
- The atomic dispersion of yttria changes the diffusion coefficients of Cr, O, and W: at one selected spot on the lamella, Figure 8, multiple grain boundaries are visible in the electron image. Research has shown yttria segregation at the grain boundaries [28]. However, here yttria is homogeneously distributed within the W matrix at atomic level. In the absence of diffusion data of yttria in W, we calculated using the data from metallic Y: the diffusion coefficient D of Y in W is given in analogy to Equation (3) with the pre-exponential factor , and the diffusion energy valid in the temperature range from to [52]. Therefore, the displacement of metallic Y atoms by diffusion during the oxidation time of would be of the same order of magnitude as the typical grain size of the alloy. However, the Y is bound to oxygen. The bond length of each bond is [53], whereas the atomic radius of an Y3+ ion is only [45]. Thus, Y2O3 is very immobile and the atomic dispersion of the yttria, originating from atomic deposition in the magnetron, is stable throughout the oxidation experiment. This dispersion can change the diffusion coefficients of Cr, O, and W.
7. Conclusions and Outlook
- A protective oxide layer forms which suppresses sublimation. Failure of the protective layer occurs in case of Cr depletion by W breaking through the protective oxide layer. Interpolation of the presented data predicts that the W-12Cr-0.5Y alloy can withstand a LOCA exposing the material to dry, O2-containing atmosphere at .
- Cr continuously diffuses to the outermost surface of the oxide, expanding freely and forming new protective oxides. Due to free expansion, stresses that could cause spallation are avoided.
- Passivation can work, even after the formation of (W, Cr) and (Cr, W) phases on a nm-scale. Cr is continuously replenished from the bulk of the sample, including the (Cr, W) phase, to the surface.
- Yttria is homogeneously distributed within the W matrix and enriched in nucleation sites at the interface between oxide and metal. This distribution is different from the classical anticipation of Y distribution in localized areas in most cases in between the grains [33]. Yttria significantly improves the oxidation resistance as binary W-11Cr samples break within minutes at
Author Contributions
Funding
Conflicts of Interest
References
- Shultis, J.; Faw, R. Fundamentals of Nuclear Science and Engineering; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Yu, I.; Bazylev, B.; Landman, I.; Fetzer, R. Design Strategy for the PFC in DEMO Reactor; KIT Scientific Publishing: Karlsruhe, Germany, 2013; Volume 7637. [Google Scholar] [CrossRef]
- Maisonnier, D.; Cook, I.; Sardain, P.; Boccaccini, L.; Bogusch, E.; Broden, K.; Di Pace, L.; Forrest, R.; Giancarli, L.; Hermsmeyer, S.; et al. The European power plant conceptual study. Fusion Eng. Des. 2005, 75, 1173–1179. [Google Scholar] [CrossRef]
- Lukac, F.; Vilemova, M.; Nevrla, B.; Klecka, J.; Chraska, T.; Molnarova, O. Properties of Mechanically Alloyed W-Ti Materials with Dual Phase Particle Dispersion. Metals 2017, 7, 3. [Google Scholar] [CrossRef]
- Koch, F.; Brinkmann, J.; Lindig, S.; Mishra, T.P.; Linsmeier, C. Oxidation behaviour of silicon-free tungsten alloys for use as the first wall material. Phys. Scr. 2011, T145, 014019. [Google Scholar] [CrossRef]
- Linsmeier, C.; Rieth, M.; Aktaa, J.; Chikada, T.; Hoffmann, A.; Hoffmann, J.; Houben, A.; Kurishita, H.; Jin, X.; Li, M.; et al. Development of advanced high heat flux and plasma-facing materials. Nucl. Fusion 2017, 57, 092007. [Google Scholar] [CrossRef] [Green Version]
- Coenen, J.W.; Antusch, S.; Aumann, M.; Biel, W.; Du, J.; Engels, J.; Heuer, S.; Houben, A.; Hoeschen, T.; Jasper, B.; et al. Materials for DEMO and reactor applications - boundary conditions and new concepts. Phys. Scr. 2016, T167, 014002. [Google Scholar] [CrossRef]
- Ueda, Y.; Schmid, K.; Balden, M.; Coenen, J.W.; Loewenhoff, T.; Ito, A.; Hasegawa, A.; Hardie, C.; Porton, M.; Gilbert, M. Baseline high heat flux and plasma facing materials for fusion. Nucl. Fusion 2017, 57, 092006. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Sublet, J.C.; Forrest, R.A. Handbook of Activation, Transmutation, and Radiation Damage Properties of the Elements Simulated Using FISPACT-II & TENDL; Culham Center For Fusion Energy: Abingdon, UK, 2015.
- Gilbert, M.R.; Eade, T.; Bachmann, C.; Fischer, U.; Taylor, N.P. Activation, decay heat, and waste classification studies of the European DEMO concept. Nucl. Fusion 2017, 57, 046015. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, Y.; Fang, Q.; Liu, R.; Xie, Z.; Zhang, T.; Wang, X.; Liu, C. Comparative Investigation of Tungsten Fibre Nets Reinforced Tungsten Composite Fabricated by Three Different Methods. Metals 2017, 7, 249. [Google Scholar] [CrossRef]
- Rieth, M.; Dudarev, S.; de Vicente, S.G.; Aktaa, J.; Ahlgren, T.; Antusch, S.; Armstrong, D.; Balden, M.; Baluc, N.; Barthe, M.F. A brief summary of the progress on the EFDA tungsten materials program. J. Nucl. Mater. 2013, 442, 173–180. [Google Scholar] [CrossRef]
- Coenen, J.W.; Mao, Y.; Almanstötterg, J.; Calvo, A.; Sistla, S.; Gietl, H.; Jasper, B.; Riesch, J.; Rieth, M.; Pintsuk, G.; et al. Advanced Materials for a Damage Resilient Divertor Concept for DEMO. Fusion Eng. Des. 2017, 124, 964–968. [Google Scholar] [CrossRef]
- Jasper, B.; Schoenen, S.; Du, J.; Hoeschen, T.; Koch, F.; Linsmeier, C.; Neu, R.; Riesch, J.; Terra, A.; Coenen, J.W. Behavior of tungsten fiber-reinforced tungsten based on single fiber push-out study. Nucl. Mater. Energy 2016, 9, 416. [Google Scholar] [CrossRef]
- Brandes, E.; Brook, G.; Paufler, P. (Eds.) Smithells Metals Reference Book; Butterworth-Heinemann: Oxford, UK, 1992. [Google Scholar]
- Wegener, T.; Klein, F.; Litnovsky, A.; Rasinski, M.; Brinkmann, J.; Koch, F.; Linsmeier, C. Development of yttrium-containing self-passivating tungsten alloys for future fusion power plants. Nucl. Mater. Energy 2016, 9, 394–398. [Google Scholar] [CrossRef]
- Litnovsky, A.; Wegener, T.; Klein, F.; Linsmeier, C.; Rasinski, M.; Kreter, A.; Unterberg, B.; Coenen, J.W.; Du, H.; Mayer, J.; et al. Smart tungsten alloys as a material for the first wall of a future fusion power plant. Nucl. Fusion 2017, 57, 066020. [Google Scholar] [CrossRef]
- Litnovsky, A.; Wegener, T.; Klein, F.; Linsmeier, C.; Rasinski, M.; Kreter, A.; Unterberg, B.; Vogel, M.; Kraus, S.; Breuer, U.; et al. Smart alloys for a future fusion power plant: First studies under stationary plasma load and in accidental conditions. Nucl. Mater. Energy 2016, 12, 1363–1367. [Google Scholar] [CrossRef]
- Eckstein, W. Sputtering by Particle Bombardment: Experiments and Computer Calculations from Threshold to MeV Energies; Chapter Sputtering Yields; Springer: Berlin/Heidelberg, Germany, 2007; pp. 33–187. [Google Scholar]
- Litnovsky, A.; Wegener, T.; Klein, F.; Linsmeier, C.; Rasinski, M.; Kreter, A.; Tan, X.Y.; Schmitz, J.; Mao, Y.; Coenen, J.W.; et al. Advanced smart tungsten alloys for a future fusion power plant. Plasma Phys. Control. Fusion 2017, 59, 064003. [Google Scholar] [CrossRef]
- Wallwork, G.R.; Hed, A.Z. Some limiting factors in the use of alloys at high temperatures. Oxid. Met. 1971, 3, 171–184. [Google Scholar] [CrossRef]
- Koch, F.; Bolt, H. Self passivating W-based alloys as plasma facing material for nuclear fusion. Phys. Scr. 2007, T128, 100–105. [Google Scholar] [CrossRef]
- Telu, S.; Mitra, R.; Pabi, S.K. Effect of Y2O3 Addition on Oxidation Behavior of W-Cr Alloys. Metall. Trans. 2015, 46, 5909–5919. [Google Scholar] [CrossRef]
- Birks, N.; Meier, G.; Pettit, F. Introduction to the High-Temperature Oxidation of Metals; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Griffiths, W.T.; Pfeil, L. Improvements in Heat Resistant Alloys. U.K. Patent No. 459848, 11 January 1937. [Google Scholar]
- Lang, E. (Ed.) The Role of Active Elements in the Oxidation Behaviour of High Temperature Metals and Alloys; Elsevier Applied Science: London, UK, 1989. [Google Scholar]
- Buergel, R.; Maier, H.J.; Niendorf, T. Handbuch Hochtemperatur-Werkstofftechnik; PRAXIS: Chichester, UK, 2011. [Google Scholar]
- Przybylski, K.; Garratt-Reed, A.J.; Yurek, G.J. Grain Boundary Segregation of Yttrium in Chromia Scales. J. Electrochem. Soc. 1988, 135, 509–517. [Google Scholar] [CrossRef]
- Stringer, J.; Wilcox, B.A.; Jaffee, R.I. The high-temperature oxidation of nickel-20 wt. % chromium alloys containing dispersed oxide phases. Oxid. Met. 1972, 5, 11–47. [Google Scholar] [CrossRef]
- Quadakkers, W.J.; Holzbrecher, H.; Briefs, K.; Beske, H. The Effect of Yttria Dispersions on the Growth Mechanisms and Morphology of Chromia and Alumina Scales. In The Role of Active Elements in the Oxidation Behaviour of High Temperature Metals and Alloys; Lang, E., Ed.; Springer: Dordrecht, The Netherlands, 1989; pp. 155–173. [Google Scholar]
- Mevrel, R. Cyclic oxidation of high-temperature alloys. J. Mater. Sci. Technol. 1987, 3, 531–535. [Google Scholar] [CrossRef]
- Tang, Q.; Sun, H.; Zhou, M.; Quan, G. Effect of Y Addition on the Semi-Solid Microstructure Evolution and the Coarsening Kinetics of SIMA AZ80 Magnesium Alloy. Metals 2017, 7, 416. [Google Scholar] [CrossRef]
- Saito, Y.; Önay, B.; Maruyama, T. The reactive element effect (REE) in oxidation of alloys. J. Phys. IV 1993, 3, 217–230. [Google Scholar] [CrossRef]
- Stroosnijder, M.; Sunderkijtter, J.D.; Cristobal, M.J.; Jenett, H.; Isenbügel, K.; Baker, M.A. The influence of yttrium ion implantation on the oxidation behaviour of powder metallurgically produced chromium. Surf. Coat. Technol. 1996, 83, 205–211. [Google Scholar] [CrossRef]
- Wegener, T.; Klein, F.; Litnovsky, A.; Rasinski, M.; Brinkmann, J.; Koch, F.; Linsmeier, C. Development and analyses of self-passivating tungsten alloys for DEMO accidental conditions. Fusion Eng. Des. 2017, 124, 183–186. [Google Scholar] [CrossRef]
- Schmitz, J.; Litnovsky, A.; Klein, F.; Wegener, T.; Tan, X.Y.; Rasinski, M.; Mutzke, A.; Hansen., P.; Kreter, A.; Pospieszczyk, A.; et al. WCrY smart alloys as advanced plasma-facing materials—Exposure to steady-state pure deuterium plasmas in PSI-2. Nucl. Mater. Energy 2018. In Press. [Google Scholar] [CrossRef]
- Ernst Ruska-Centre for Microscopy and Spectroscopy with Electrons. FEI Titan 80-300 STEM. JLSRF 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Turkdogan, E. Physical Chemistry of High Temperature Technology; Academic Press: New York, NY, USA, 1980. [Google Scholar]
- Calvo, A.; Ordas, N.; Iturriza, I.; Pastor, J.Y.; Tejado, E.; Palacios, T.; Garcia-Rosales, C. Manufacturing of self-passivating tungsten based alloys by different powder metallurgical routes. Phys. Scr. 2016, T167, 014041. [Google Scholar] [CrossRef]
- Swanson, H.E. Standard X-ray Diffraction Powder Patterns; National Bureau of Standards: Washington, DC, USA, 1953; Volume 1, p. 28.
- Thornton, J.A. Influence of substrate temperature and deposition rate on structure of thick sputtered Cu coatings. J. Vac. Sci. Technol. 1975, 12, 830. [Google Scholar] [CrossRef]
- Ross, R.G.; Hume-Rothery, W. High temperature X-ray metallography: I. A new debye-scherrer camera for use at very high temperatures II. A new parafocusing camera III. Applications to the study of chromium, hafnium, molybdenum, rhodium, ruthenium and tungsten. J. Less-Common Met. 1963, 5, 258–270. [Google Scholar] [CrossRef]
- Reed, T.B. (Ed.) Free Energy of Formation of Binary Compounds; MIT Press: Cambridge, MA, USA, 1971. [Google Scholar]
- Royer, L.; Ledoux, X.; Mathieu, S.; Steinmetz, P. On the Oxidation and Nitridation of Chromium at 1300 C. Oxid. Met. 2010, 74, 79–92. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef] [Green Version]
- Litnovsky, A.; Wegener, T.; Klein, F.; Linsmeier, C.; Rasinski, M.; Kreter, A.; Tan, X.Y.; Schmitz, J.; Coenen, J.W.; Mao, Y.; et al. New oxidation-resistant tungsten alloys for use in the nuclear fusion reactors. Phys. Scr. 2017, T170, 014012. [Google Scholar] [CrossRef]
- Rosman, K.J.R.; Taylor, P.D.P. Isotopic compositions of the elements 1997. Pure Appl. Chem. 1998, 70, 217–235. [Google Scholar] [CrossRef]
- Thompson, A.C.; Attwood, D.T.; Gullikson, E.M.; Howells, M.R.; Kortright, J.B.; Robinson, A.L.; Underwood, J.H.; Kim, K.; Kirz, J.; Lindau, I.; et al. X-ray Data Booklet; Center for X-ray Optics and Advanced Light Source; Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2001.
- Andersen, C.A. Progress in analytic methods for the ion microprobe mass analyzer. Int. J. Mass Spectrom. Ion Phys. 1969, 2, 61. [Google Scholar] [CrossRef]
- Klotsman, S.M.; Koloskov, V.; Osetrov, S.; Polikarpova, I.; Tatarinova, G.; Timofeev, A. Chromium and molybdenum diffusion in tungsten single crystals. Fiz. Met. Metalloved. 1989, 67, 767–774. [Google Scholar]
- Lee, J.; Klockgeter, K.; Herzig, C. Grain boundary self and impurity diffusion in Tungsten in the Temperature range of activated sintering. J. Phys. Colloq. 1990, 51, 569–574. [Google Scholar] [CrossRef]
- Gornyy, D.; Altovskiy, R. Diffusion of Yt in refractory metals. Fiz. Met. Metalloved. 1971, 31, 781. [Google Scholar]
- Lazdins, K.; Kuzmin, A. Local structure and lattice dynamics of cubic Y2O3: An X-ray absorption spectroscopy study. In Proceedings of the OP Conference Series: Materials Science and Engineering, St George, UT, USA, 6–10 November 2017; Volume 77. [Google Scholar] [CrossRef]
- Zhu, G.; Libby, C. Review and Future Perspective of Central Receiver Design and Performance. AIP Conf. Proc. 2017, 1850. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, F.; Wegener, T.; Litnovsky, A.; Rasinski, M.; Tan, X.; Schmitz, J.; Linsmeier, C.; Coenen, J.W.; Du, H.; Mayer, J.; et al. On Oxidation Resistance Mechanisms at 1273 K of Tungsten-Based Alloys Containing Chromium and Yttria. Metals 2018, 8, 488. https://doi.org/10.3390/met8070488
Klein F, Wegener T, Litnovsky A, Rasinski M, Tan X, Schmitz J, Linsmeier C, Coenen JW, Du H, Mayer J, et al. On Oxidation Resistance Mechanisms at 1273 K of Tungsten-Based Alloys Containing Chromium and Yttria. Metals. 2018; 8(7):488. https://doi.org/10.3390/met8070488
Chicago/Turabian StyleKlein, Felix, Tobias Wegener, Andrey Litnovsky, Marcin Rasinski, Xiaoyue Tan, Janina Schmitz, Christian Linsmeier, Jan Willem Coenen, Hongchu Du, Joachim Mayer, and et al. 2018. "On Oxidation Resistance Mechanisms at 1273 K of Tungsten-Based Alloys Containing Chromium and Yttria" Metals 8, no. 7: 488. https://doi.org/10.3390/met8070488
APA StyleKlein, F., Wegener, T., Litnovsky, A., Rasinski, M., Tan, X., Schmitz, J., Linsmeier, C., Coenen, J. W., Du, H., Mayer, J., & Breuer, U. (2018). On Oxidation Resistance Mechanisms at 1273 K of Tungsten-Based Alloys Containing Chromium and Yttria. Metals, 8(7), 488. https://doi.org/10.3390/met8070488