
Complete Genomic RNA Sequence of Tuberose Mild Mosaic Virus and Tuberose Mild Mottle Virus Acquired by High-Throughput Sequencing

 ,
,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
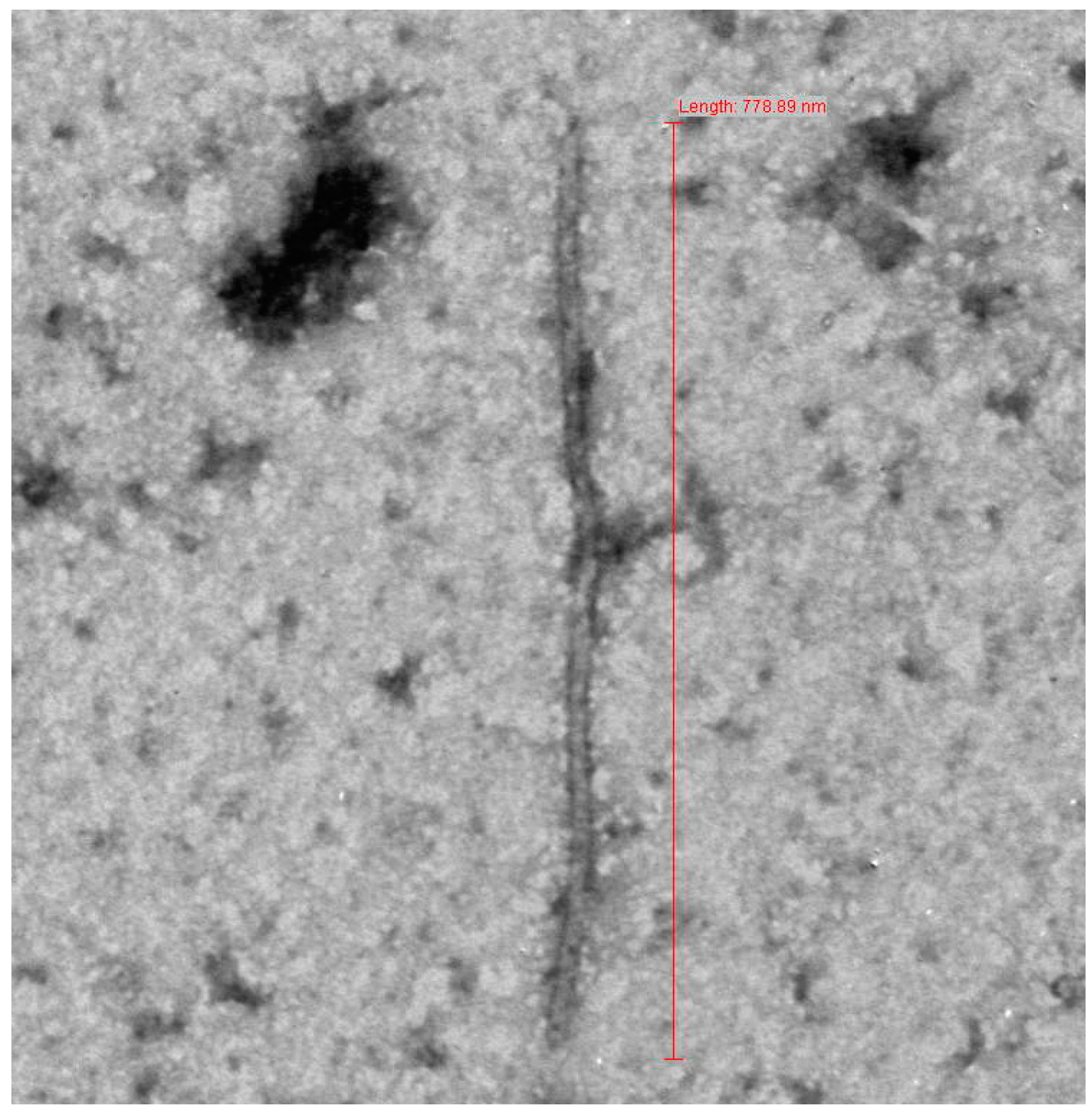
3.1. Sample Collection and Electron Microscopy
3.2. RNA Extraction, Library Preparation and HTS
3.3. Data Processing and Virus Analysis
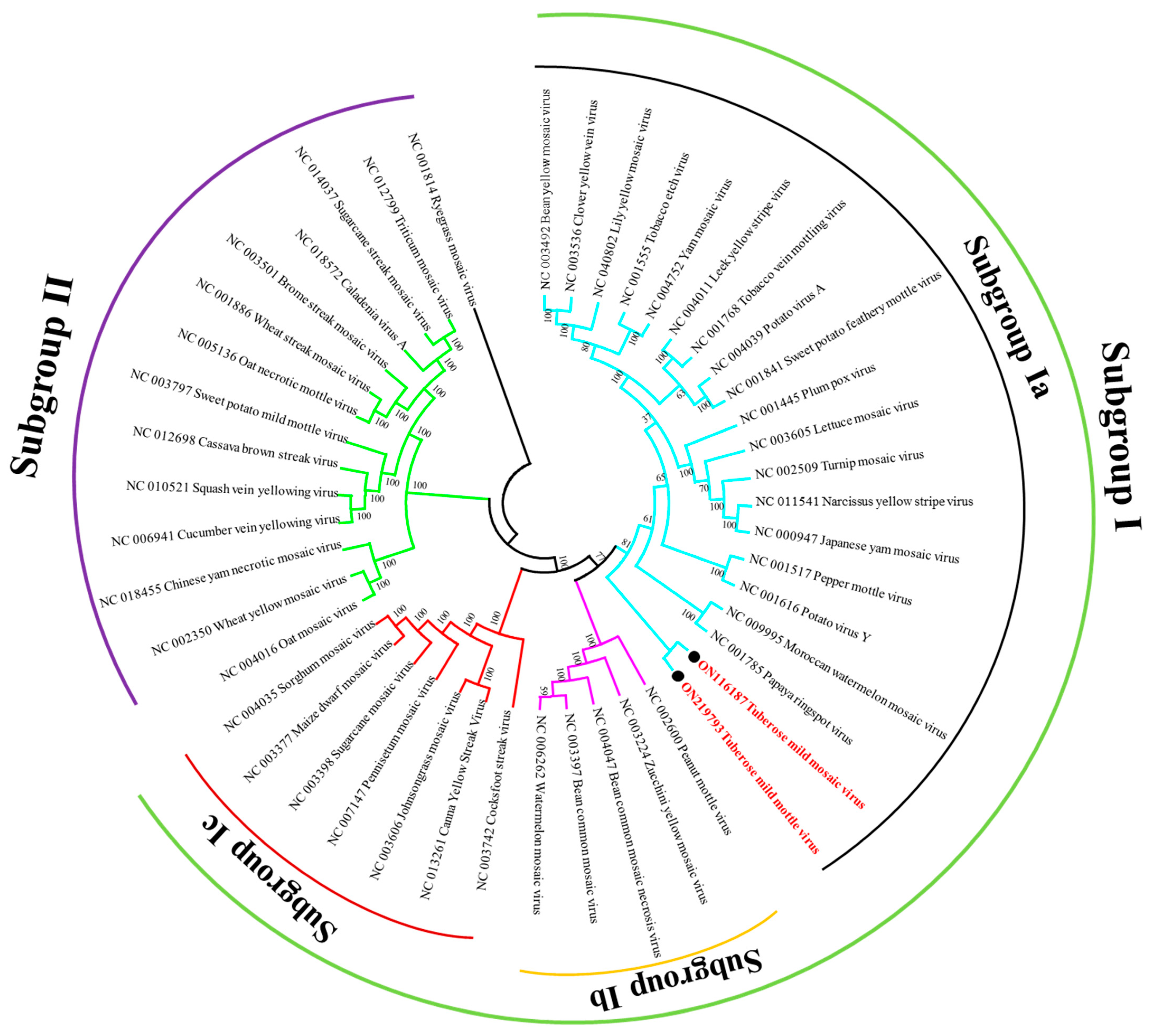
3.4. Sequence Similarity and Phylogenetic Analysis
3.5. RT-PCR and Sanger Sequencing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verma, C.; Kashyap, S.; Kerketta, A. Major Diseases of Tuberose and Their Management. Pop. Kheti 2020, 8, 74–76. [Google Scholar]
- Pasin, F.; Daròs, J.A.; Tzanetakis, I.E. Proteome expansion in the Potyviridae evolutionary radiation. FEMS Microbiol. Rev. 2022, 46, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Inoue-Nagata, A.K.; Jordan, R.; Kreuze, J.; Li, F.; López-Moya, J.J.; Mäkinen, K.; Ohshima, K.; Wylie, S.J. ICTV Virus Taxonomy Profile: Potyviridae. J. Gen. Virol. 2022, 103, 001738. [Google Scholar]
- Wylie, S.J.; Adams, M.; Chalam, C.; Kreuze, J.; López-Moya, J.J.; Ohshima, K.; Praveen, S.; Rabenstein, F.; Stenger, D.; Wang, A.; et al. ICTV virus taxonomy profile: Potyviridae. J. Gen. Virol. 2017, 98, 352. [Google Scholar] [CrossRef]
- Simmons, H.E.; Dunham, J.P.; Zinn, K.E.; Munkvold, G.P.; Holmes, E.C.; Stephenson, A.G. Zucchini yellow mosaic virus (ZYMV, Potyvirus): Vertical transmission, seed infection and cryptic infections. Virus Res. 2013, 176, 259–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadidi, A.; Flores, R.; Candresse, T.; Barba, M. Next-generation sequencing and genome editing in plant virology. Front. Microbiol. 2016, 7, 1325. [Google Scholar] [CrossRef] [PubMed]
- Pecman, A.; Kutnjak, D.; Gutiérrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next generation sequencing for detection and discovery of plant viruses and viroids: Comparison of two approaches. Front. Microbiol. 2017, 8, 1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidharthan, V.K.; Kalaivanan, N.S.; Baranwal, V.K. Discovery of putative novel viruses in the transcriptomes of endangered plant species native to India and China. Gene 2021, 786, 145626. [Google Scholar] [CrossRef]
- Prajapati, M.R.; Manav, A.; Singh, J.; Singh, M.K.; Ranjan, K.; Kumar, A.; Kumar, P.; Kumar, R.; Baranwal, V.K. Identification of Garlic virus A infecting Allium sativum L. through next generation sequencing technology. J. Hortic. Sci. Biotechnol. 2022, 97, 96–105. [Google Scholar] [CrossRef]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High throughput sequencing for plant virus detection and discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Truong, T.N.; An, D.; Prajapati, M.R.; Manav, A.; Quoc, N.B.; Ranjan, K.; Kumar, A.; Kumar, P.; Kumar, R.; et al. Complete genome sequence and genetic organization of a Garlic virus D infecting garlic (Allium sativum) from northern India. Acta Virol. 2020, 64, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, M.R.; Manav, A.; Singh, J.; Kumar, P.; Kumar, A.; Kumar, R.; Prakash, S.; Baranwal, V.K. Identification and Characterization of a Garlic Virus E Genome in Garlic (Allium sativum L.) Using High-Throughput Sequencing from India. Plants 2022, 11, 224. [Google Scholar] [CrossRef]
- Kainthura, P.; Srivastava, R. Induction of genetic variability and isolation of mutants in tuberose (Polianthes tuberosa L.). Trop. Agric. Res. 2015, 26, 721–732. [Google Scholar] [CrossRef] [Green Version]
- Harrison, B.D.; Finch, J.T.; Gibbs, A.J.; Hollings, M.; Shepherd, R.J.; Valenta, V.; Wetter, C. Sixteen groups of plant viruses. Virology 1971, 45, 356–363. [Google Scholar] [CrossRef]
- Adams, I.P.; Miano, D.W.; Kinyua, Z.M.; Wangai, A.; Kimani, E.; Phiri, N.; Reeder, R.; Harju, V.; Glover, R.; Hany, U.; et al. Use of next-generation sequencing for the identification and characterization of M aize chlorotic mottle virus and S ugarcane mosaic virus causing maize lethal necrosis in Kenya. Plant Pathol. 2013, 62, 741–749. [Google Scholar] [CrossRef]
- Perotto, M.C.; Pozzi, E.A.; Celli, M.G.; Luciani, C.E.; Mitidieri, M.S.; Conci, V.C. Identification and characterization of a new potyvirus infecting cucurbits. Arch. Virol. 2018, 163, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, S.; Mehra, A.; Hallan, V.; Raikhy, G.; Ram, R.; Zaidi, A.A. Molecular evidence for occurrence of Tuberose mild mottle virus infecting tuberose (Polianthes tuberosa) in India. Curr. Sci. 2005, 89, 870–872. [Google Scholar]
- Raj, S.K.; Snehi, S.K.; Kumar, S.; Ram, T.; Goel, A.K. First report of Tuberose mild mosaic potyvirus from tuberose (Polianthes tuberosa L.) in India. Australas. Plant Dis. Notes 2009, 4, 93–95. [Google Scholar]
- Dey, K.K.; Melzer, M.J.; Li, C.; Sun, X.; Adkins, S. First report of tuberose mild mottle virus infecting tuberose (Polianthes tuberosa) in the United States. Plant Disease 2018, 102, 461. [Google Scholar] [CrossRef]
- Lin, L.; Zheng, H.Y.; Chen, J.; Chen, J.P.; Zhang, Q.Y.; Zhao, M.F.; Antoniw, J.F.; Adams, M.J. A new potyvirus from tuberose (Polianthes tuberosa) in China. Arch. Virol. 2004, 149, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Lucinda, N.; Da Rocha, W.B.; Inoue-Nagata, A.K.; Nagata, T. Complete genome sequence of pepper yellow mosaic virus, a potyvirus, occurring in Brazil. Arch. Virol. 2012, 157, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Knierim, D.; Menzel, W.; Winter, S. Analysis of the complete genome sequence of euphorbia ringspot virus, an atypical member of the genus Potyvirus. Arch. Virol. 2017, 162, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.Y.W.; Miller, W.A.; Atkins, J.F. & Firth, A.E. An overlapping essential gene in the Potyviridae. Proc. Natl. Acad. Sci. USA 2008, 105, 5897–5902. [Google Scholar] [PubMed] [Green Version]
- Olspert, A.; Chung, B.Y.W.; Atkins, J.F.; Carr, J.P.; Firth, A.E. Transcriptional slippage in the positive-sense RNA virus family Potyviridae. EMBO Rep. 2015, 16, 995–1004. [Google Scholar] [CrossRef]
- Nigam, D.; LaTourrette, K.; Souza, P.F.; Garcia-Ruiz, H. Genome-wide variation in potyviruses. Front. Plant Sci. 2019, 10, 1439. [Google Scholar] [CrossRef] [Green Version]
- Lan, P.; He, P.; Zhang, Y.; Zhang, S.; Zhang, Z.; Chen, X.; Tan, S.; Luo, H.; Cao, M.; Li, F. Molecular characterization of a novel potyvirus infecting noni. Arch. Virol. 2019, 164, 3099–3102. [Google Scholar] [CrossRef] [PubMed]
- Worrall, E.A.; Hayward, A.C.; Fletcher, S.J.; Mitter, N. Molecular characterization and analysis of conserved potyviral motifs in bean common mosaic virus (BCMV) for RNAi-mediated protection. Arch. Virol. 2019, 164, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Revers, F.; García, J.A. Molecular biology of potyviruses. Adv. Virus Res. 2015, 92, 101–199. [Google Scholar] [PubMed]
- Gibbs, A.J.; Mackenzie, A.M.; Wei, K.J.; Gibbs, M.J. The potyviruses of Australia. Arch. Virol. 2008, 153, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, Y.; Nie, X.; Giguère, M.A.; Nanayakkara, U.; Maw, E.; Foottit, R. A new approach for the identification of aphid vectors (Hemiptera: Aphididae) of potato virus Y. J. Econ. Entomol. 2012, 105, 1909–1914. [Google Scholar] [CrossRef]
- Romay, G.; Lecoq, H.; Desbiez, C. Zucchini tigré mosaic virus is a distinct potyvirus in the papaya ringspot virus cluster: Molecular and biological insights. Arch. Virol. 2014, 159, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Manav, A.; Prajapati, M.R.; Singh, J.; Kumar, A.; Kumar, P.; Pant, R.P.; Baranwal, V.K. First report of natural infection by two potyviruses on amaryllis (Hippeastrum hybridum) plants from India. VirusDisease 2021, 32, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit Sequence Alignment Editor 7.0. 1; Isis Pharmaceuticals: Carlsbad, CA, USA, 2004. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sequence Identity nts/aa | ON116187 Tuberose mild mosaic | ON219793 Tuberose mild mottle virus | NC001785 Papaya ringspot virus | NC009995 Moroccan watermelon mosaic virus | NC001616 Potato virus Y | NC001517 Pepper mottle viru | NC000947 Japanese yam mosaic virus | NC011541 Narcissus yellow stripe virus | NC002509 Turnip mosaic virus | NC003605 Lettuce mosaic virus | NC001445 Plum pox virus | NC001841 Sweet potato feathery mottle virus | NC004039 Potato virus A | NC001768 Tobacco vein mottling virus | NC004011 Leek yellow stripe virus | NC004752 Yam mosaic virus | NC001555 Tobacco etch virus | NC040802 Lily yellow mosaic virus | NC003536 Clover yellow vein virus | NC003492 Bean yellow mosaic virus |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ON116187 Tuberose mild mosaic virus | ID | 71.0 | 61.8 | 46.0 | 43.0 | 43.3 | 45.4 | 45.1 | 45.4 | 41.7 | 45.7 | 42.8 | 43.6 | 43.7 | 41.3 | 44.5 | 43.4 | 42.6 | 42.0 | 43.2 |
| ON219793 Tuberose mild mottle virus | 65.2 | ID | 62.3 | 46.2 | 43.1 | 43.3 | 44.4 | 44.9 | 45.0 | 42.0 | 45.1 | 42.3 | 43.7 | 43.9 | 41.7 | 43.6 | 43.3 | 43.0 | 43.0 | 43.5 |
| NC001785 Papaya ringspot virus | 58.1 | 57.9 | ID | 57.9 | 41.0 | 41.0 | 42.8 | 42.3 | 42.5 | 43.0 | 44.0 | 42.3 | 41.9 | 41.3 | 44.0 | 42.2 | 49.0 | 42.3 | 47.0 | 41.3 |
| NC009995 Moroccan watermelon mosaic virus | 49.5 | 49.0 | 61.0 | ID | 43.8 | 43.7 | 45.0 | 45.3 | 45.8 | 43.8 | 47.6 | 44.3 | 43.8 | 44.1 | 42.9 | 45.8 | 44.3 | 44.1 | 43.9 | 44.5 |
| NC001616 Potato virus Y | 54.0 | 55.0 | 48.1 | 49.1 | ID | 61.3 | 44.8 | 45.3 | 45.9 | 45.6 | 48.0 | 43.5 | 44.7 | 45.2 | 42.0 | 47.2 | 45.7 | 43.2 | 42.8 | 43.8 |
| NC001517 Pepper mottle virus | 59.0 | 59.0 | 47.8 | 48.5 | 61.7 | ID | 45.2 | 45.7 | 46.3 | 45.0 | 47.6 | 43.6 | 44.6 | 44.3 | 42.1 | 46.3 | 45.8 | 43.3 | 43.4 | 43.7 |
| NC000947 Japanese yam mosaic virus | 50.0 | 48.8 | 49.7 | 49.9 | 49.5 | 49.9 | ID | 58.8 | 58.6 | 46.9 | 50.1 | 46.9 | 47.0 | 46.8 | 43.4 | 48.1 | 46.3 | 45.2 | 45.2 | 46.1 |
| NC011541 Narcissus yellow stripe virus | 49.2 | 48.3 | 48.7 | 49.3 | 49.8 | 49.2 | 61.9 | ID | 60.8 | 46.3 | 50.8 | 47.6 | 47.5 | 46.8 | 43.7 | 47.3 | 46.4 | 45.6 | 45.9 | 45.9 |
| NC002509 Turnip mosaic virus | 49.2 | 48.6 | 49.0 | 51.0 | 49.9 | 49.9 | 62.0 | 63.0 | ID | 47.4 | 50.9 | 49.3 | 48.5 | 46.8 | 44.1 | 47.8 | 46.8 | 45.3 | 45.2 | 45.1 |
| NC003605 Lettuce mosaic virus | 47.9 | 47.4 | 49.0 | 49.2 | 49.3 | 48.4 | 54.1 | 53.5 | 54.1 | ID | 47.7 | 45.3 | 44.1 | 43.9 | 42.4 | 46.1 | 43.7 | 42.7 | 43.0 | 42.9 |
| NC001445 Plum pox virus | 59.0 | 50.0 | 51.0 | 51.2 | 51.1 | 59.0 | 52.9 | 51.7 | 52.5 | 51.4 | ID | 50.3 | 49.1 | 47.0 | 46.2 | 50.3 | 47.3 | 47.8 | 46.4 | 47.4 |
| NC001841 Sweet potato feathery mottle virus | 48.9 | 48.9 | 54.0 | 56.0 | 49.1 | 49.7 | 51.4 | 51.4 | 51.1 | 54.0 | 54.2 | ID | 45.6 | 43.6 | 42.4 | 46.0 | 44.2 | 43.6 | 42.9 | 44.0 |
| NC004039 Potato virus A | 48.4 | 49.3 | 48.5 | 49.4 | 49.1 | 48.5 | 58.0 | 51.3 | 51.1 | 49.6 | 52.2 | 55.8 | ID | 54.6 | 43.5 | 46.9 | 48.5 | 45.8 | 45.5 | 46.4 |
| NC001768 Tobacco vein mottling virus | 49.6 | 49.4 | 47.8 | 48.3 | 53.0 | 49.2 | 49.1 | 49.1 | 48.7 | 48.9 | 49.8 | 49.6 | 52.1 | ID | 43.9 | 46.7 | 49.3 | 45.4 | 44.8 | 44.9 |
| NC004011 Leek yellow stripe virus | 49.6 | 49.2 | 48.7 | 48.5 | 49.9 | 49.7 | 49.4 | 48.6 | 49.3 | 48.5 | 51.1 | 49.6 | 49.3 | 54.6 | ID | 44.0 | 43.2 | 47.6 | 45.0 | 45.7 |
| NC004752 Yam mosaic virus | 48.8 | 47.9 | 49.0 | 49.5 | 49.1 | 49.0 | 57.0 | 49.4 | 54.0 | 49.3 | 52.0 | 51.5 | 51.3 | 48.9 | 48.5 | ID | 45.4 | 44.9 | 45.7 | 45.1 |
| NC001555 Tobacco etch virus | 48.4 | 47.7 | 47.6 | 48.6 | 49.3 | 48.7 | 49.8 | 49.3 | 49.7 | 48.6 | 51.2 | 58.0 | 51.6 | 49.8 | 48.9 | 54.8 | ID | 44.8 | 45.2 | 45.6 |
| NC040802 Lily yellow mosaic virus | 48.7 | 48.1 | 49.0 | 49.5 | 48.8 | 49.3 | 52.0 | 49.4 | 49.9 | 49.3 | 52.6 | 51.0 | 58.0 | 48.3 | 53.0 | 52.0 | 51.2 | ID | 49.3 | 49.4 |
| NC003536 Clover yellow vein virus | 48.8 | 48.2 | 48.7 | 49.4 | 48.6 | 49.2 | 50.0 | 49.5 | 56.0 | 48.6 | 51.4 | 53.0 | 58.0 | 48.5 | 49.7 | 51.9 | 52.2 | 55.8 | ID | 68.9 |
| NC003492 Bean yellow mosaic virus | 48.3 | 48.1 | 49.0 | 49.1 | 49.3 | 49.4 | 49.8 | 49.6 | 49.6 | 48.7 | 51.4 | 53.0 | 59.0 | 48.6 | 53.0 | 52.1 | 51.7 | 54.9 | 65.9 | ID |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prajapati, M.R.; Manav, A.; Singhal, P.; Sidharthan, V.K.; Sirohi, U.; Kumar, M.; Bharti, M.K.; Singh, J.; Kumar, P.; Kumar, R.; et al. Complete Genomic RNA Sequence of Tuberose Mild Mosaic Virus and Tuberose Mild Mottle Virus Acquired by High-Throughput Sequencing. Pathogens 2022, 11, 861. https://doi.org/10.3390/pathogens11080861
Prajapati MR, Manav A, Singhal P, Sidharthan VK, Sirohi U, Kumar M, Bharti MK, Singh J, Kumar P, Kumar R, et al. Complete Genomic RNA Sequence of Tuberose Mild Mosaic Virus and Tuberose Mild Mottle Virus Acquired by High-Throughput Sequencing. Pathogens. 2022; 11(8):861. https://doi.org/10.3390/pathogens11080861
Chicago/Turabian StylePrajapati, Malyaj R., Aakansha Manav, Pankhuri Singhal, Venkidusamy K. Sidharthan, Ujjwal Sirohi, Mukesh Kumar, Mahesh Kumar Bharti, Jitender Singh, Pankaj Kumar, Ravindra Kumar, and et al. 2022. "Complete Genomic RNA Sequence of Tuberose Mild Mosaic Virus and Tuberose Mild Mottle Virus Acquired by High-Throughput Sequencing" Pathogens 11, no. 8: 861. https://doi.org/10.3390/pathogens11080861
APA StylePrajapati, M. R., Manav, A., Singhal, P., Sidharthan, V. K., Sirohi, U., Kumar, M., Bharti, M. K., Singh, J., Kumar, P., Kumar, R., Prakash, S., & Baranwal, V. K. (2022). Complete Genomic RNA Sequence of Tuberose Mild Mosaic Virus and Tuberose Mild Mottle Virus Acquired by High-Throughput Sequencing. Pathogens, 11(8), 861. https://doi.org/10.3390/pathogens11080861