
Insights into the Genetic Diversity of Leishmania (Viannia) panamensis in Panama, Inferred via Multilocus Sequence Typing (MLST)

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Leishmania Isolates
2.2. Parasite Culture and DNA Extraction
2.3. Molecular Identification of Leishmania panamensis Isolates
2.3.1. PCR Amplification
2.3.2. Sanger Sequencing and Species Discrimination via Phylogenetic Analysis
2.4. PCR Amplification and Sequencing of Gene Loci Used in the MLST Approach
2.5. Sequence Assembling, Haplotype Construction, and Determination of Diversity Indexes
2.6. Determination of L. panamensis DSTs and Their Geographical Distribution
2.7. Phylogenetic Analyzes of L. panamensis DSTs
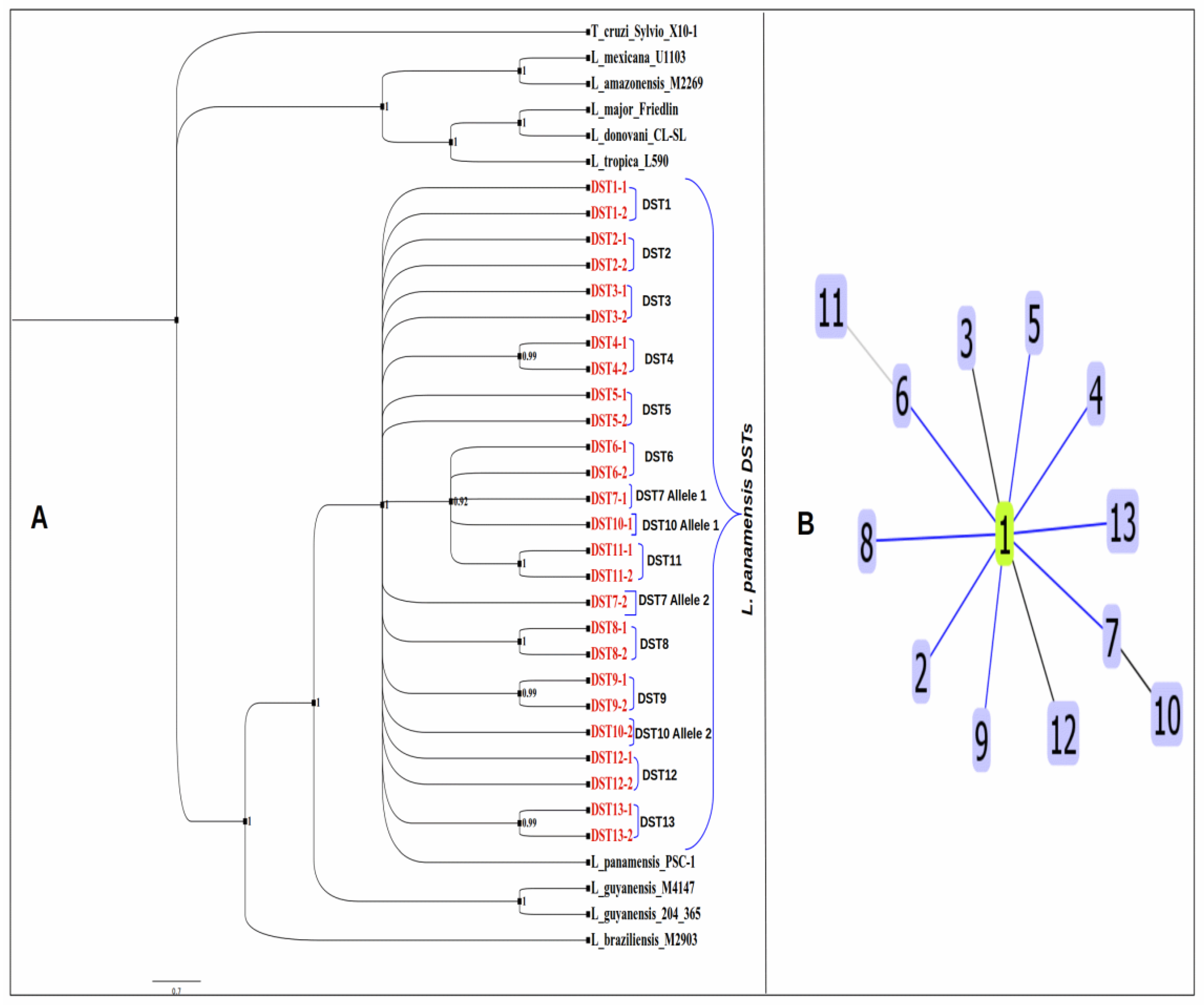
2.8. Clonal Complex Determination
2.9. Global Ancestry of Local Leishmania panamensis Isolates
3. Results
3.1. Identification of Leishmania panamensis Isolates
3.2. Genetic Diversity of L. panamensis Haplotypes
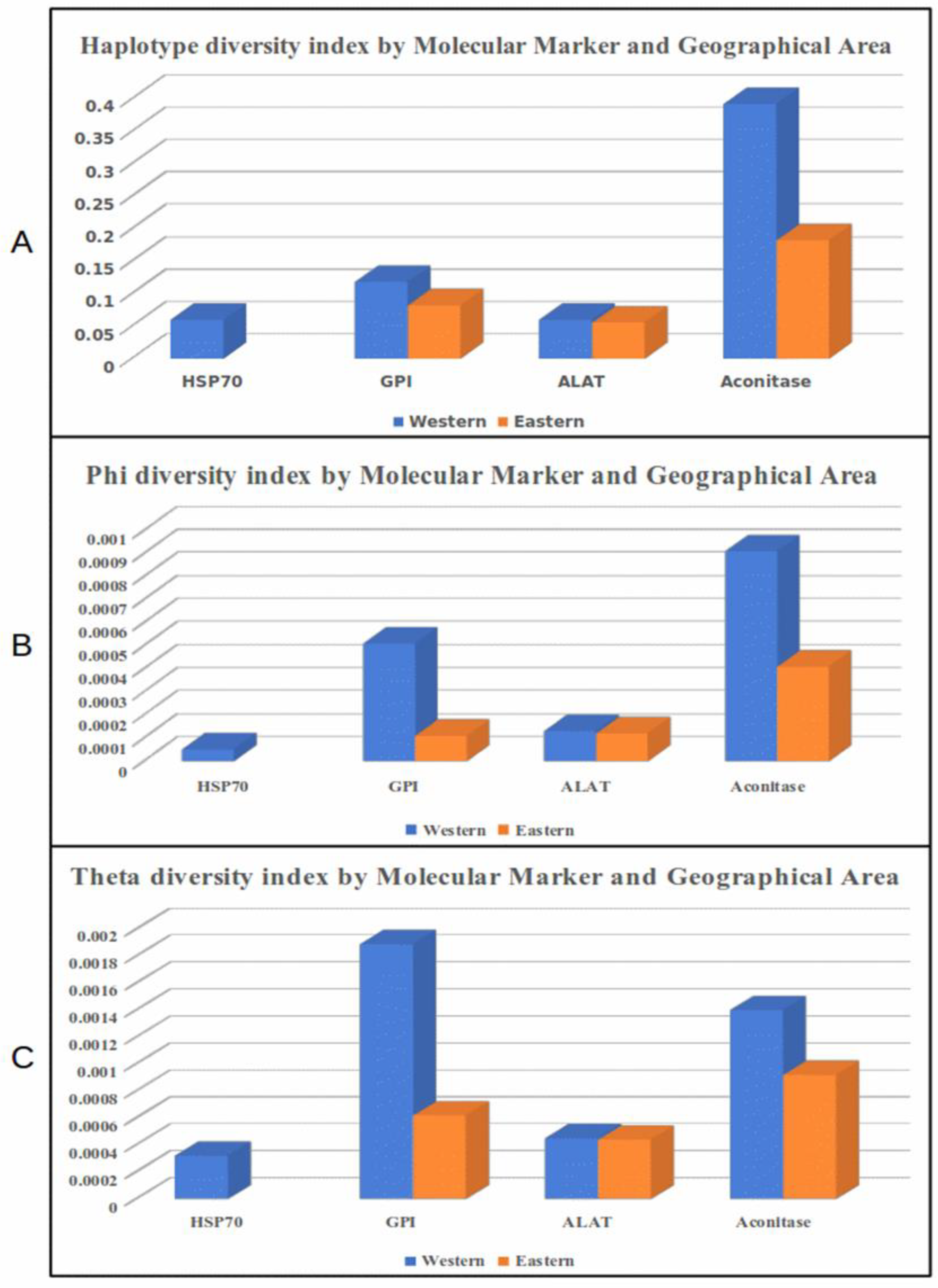
3.3. Diversity Indexes by Locus and Geographical Region
3.4. Genotyping Approach by MLST
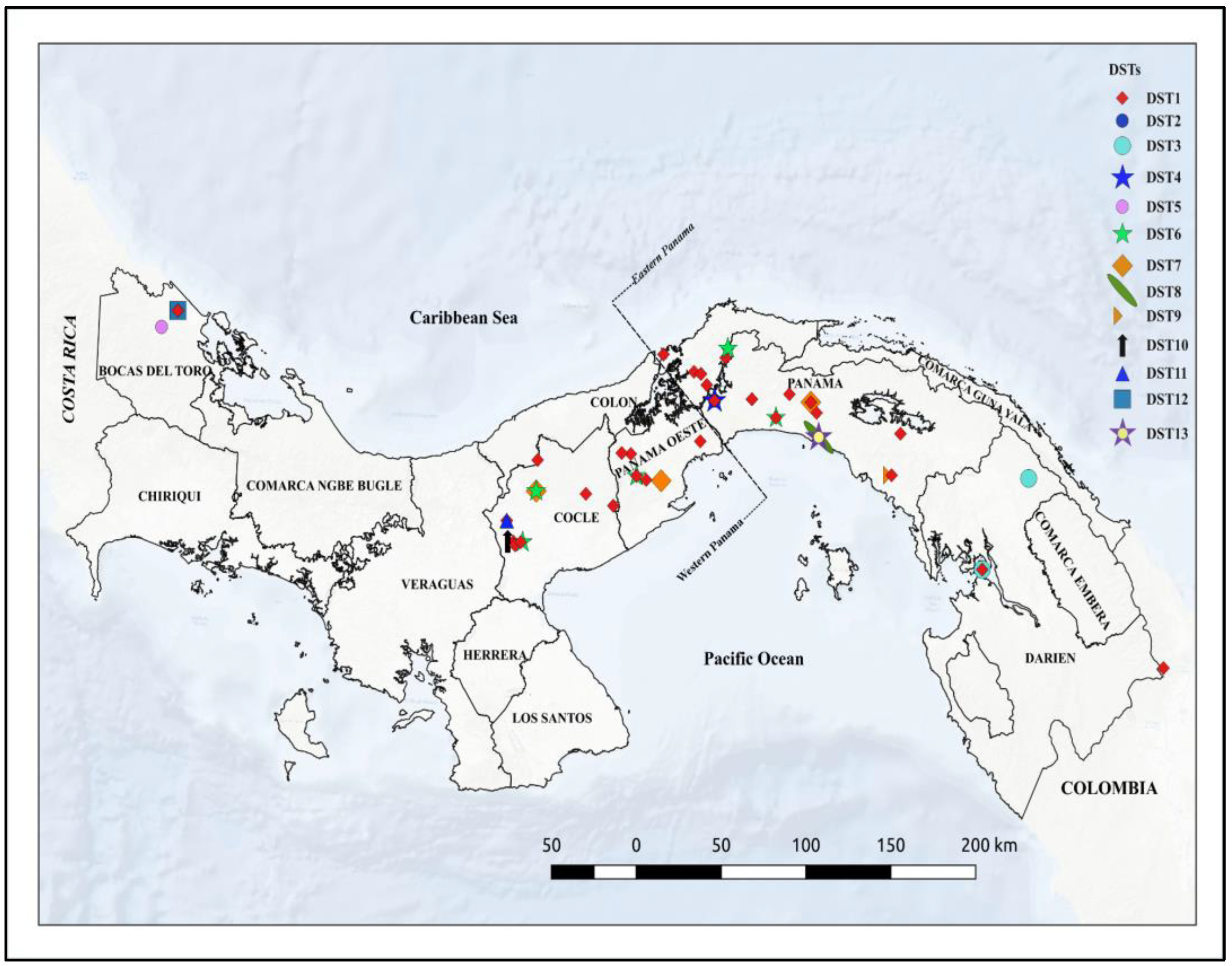
3.5. Geographic Distribution of DSTs
3.6. Haplotype Resolution of Local L. panamensis Isolates
3.7. Phylogenetic Analysis and Clonal Complex Determination of L. panamensis DSTs
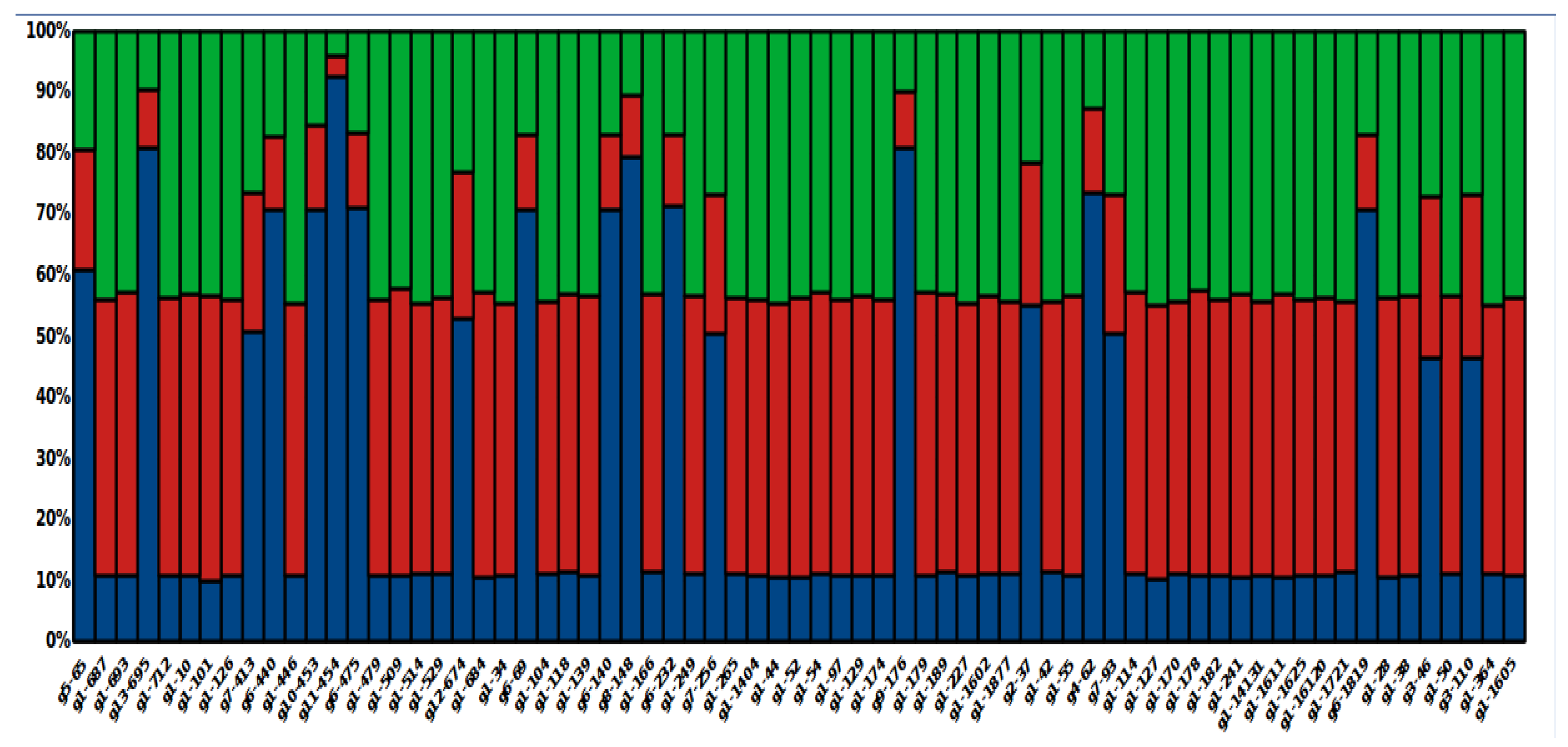
3.8. Global Ancestry of Local L. panamensis Isolates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- de Moura, T.R.; Oliveira, F.; Novais, F.O.; Miranda, J.C.; Clarêncio, J.; Follador, I.; Carvalho, E.M.; Valenzuela, J.G.; Barral-Netto, M.; Barral, A.; et al. Enhanced Leishmania braziliensis infection following pre-exposure to sandfly saliva. PLoS Negl. Trop. Dis. 2007, 1, e84. [Google Scholar] [CrossRef] [PubMed]
- Vanloubbeeck, Y.; Jones, D.E. The immunology of Leishmania infection and the implications for vaccine development. Ann. N. Y. Acad. Sci. 2004, 1026, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Pan American Health Organization. Leishmaniasis: Epidemiological Report in the Americas; Pan American Health Organization: Washington, DC, USA, 2019; pp. 2–3. [Google Scholar]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; de Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Schönian, G.; Mauricio, I.; Cupolillo, E. Is it time to revise the nomenclature of Leishmania? Trends Parasitol. 2010, 26, 466–469. [Google Scholar] [CrossRef]
- Christensen, H.A.; De Vasquez, A.M.; Petersen, J.L. Short report: Epidemiologic studies on cutaneous leishmaniasis in eastern Panama. Am. J. Trop. Med. Hyg. 1999, 60, 54–57. [Google Scholar] [CrossRef] [Green Version]
- Miranda, A.; Carrasco, R.; Paz, H.; Pascale, J.M.; Samudio, F.; Saldaña, A.; Santamaría, G.; Mendoza, Y.; Calzada, J.E. Molecular epidemiology of American tegumentary leishmaniasis in Panama. Am. J. Trop. Med. Hyg. 2009, 81, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Del Miranda, A.C.; González, K.A.; Samudio, F.; Pineda, V.J.; Calzada, J.E.; Capitan-Barrios, Z.; Jiménez, A.; Castillo, J.; Mendoza, Y.; Suárez, J.A.; et al. Molecular identification of parasites causing cutaneous leishmaniasis in Panama. Am. J. Trop. Med. Hyg. 2021, 104, 1326–1334. [Google Scholar] [CrossRef]
- Vasquez, A.N.A.M.D.E.; Saenz, R.E.; Petersen, J.L.; Johnson, C.M. Leishmania Mexicana complex: Human infections in the Republic of Panama. Am. J. Trop. Med. Hyg. 1990, 43, 619–622. [Google Scholar] [CrossRef]
- Miranda, A.; Samudio, F.; Saldaña, A.; Castillo, J.; Brandão, A.; Calzada, J.E. The calmodulin intergenic spacer as molecular target for characterization of Leishmania species. Parasites Vectors 2014, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Miranda, A.; Samudio, F.; González, K.; Saldaña, A.; Brandão, A.; Calzada, J.E. Calmodulin polymerase chain reaction-restriction fragment length polymorphism for leishmania identification and typing. Am. J. Trop. Med. Hyg. 2016, 95, 383–387. [Google Scholar] [CrossRef] [Green Version]
- Davila, M.; Pineda, V.; Calzada, J.E.; Saldaña, A.; Samudio, F. Evaluation of cytochrome b sequence to identify Leishmania species and variants: The case of Panama. Mem. Inst. Oswaldo Cruz 2021, 116, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, Y.; Gomez, E.L.; Kato, H.; Martini, L.R.; Velez, L.N.; Uezato, H. Diffuse and disseminated cutaneous leishmaniasis: Clinical cases experienced in Ecuador and a brief review. Trop. Med. Health 2016, 44, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osorio, L.E.; Castillo, C.M.; Ochoa, M.T. Mucosal Leishmaniasis due to Leishmania (Viannia) panamensis in Colombia: Clinical characteristics. Am. J. Trop. Med. Hyg. 1998, 59, 49–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freites, C.O.; Gundacker, N.D.; Pascale, J.M.; Saldaña, A.; Diaz-Suarez, R.; Jimenez, G.; Sosa, N.; García, E.; Jimenez, A.; Suarez, J.A. First case of diffuse leishmaniasis associated with leishmania panamensis. Open Forum Infect. Dis. 2018, 5, ofy281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldi, G.; Tesh, R.B.; McMahon-Pratt, D. A review of the geographic distribution and epidemiology of leishmaniasis in the New World. Am. J. Trop. Med. Hyg. 1989, 41, 687–725. [Google Scholar] [CrossRef]
- Mauricio, I.L.; Yeo, M.; Baghaei, M.; Doto, D.; Pratlong, F.; Zemanova, E.; Dedet, J.P.; Lukes, J.; Miles, M.A. Towards multilocus sequence typing of the Leishmania donovani complex: Resolving genotypes and haplotypes for five polymorphic metabolic enzymes (ASAT, GPI, NH1, NH2, PGD). Int. J. Parasitol. 2006, 36, 757–769. [Google Scholar] [CrossRef]
- Zemanová, E.; Jirků, M.; Mauricio, I.L.; Horák, A.; Miles, M.A.; Lukeš, J. The Leishmania donovani complex: Genotypes of five metabolic enzymes (ICD, ME, MPI, G6PDH, and FH), new targets for multilocus sequence typing. Int. J. Parasitol. 2007, 37, 149–160. [Google Scholar] [CrossRef]
- Boité, M.C.; Mauricio, I.L.; Miles, M.A.; Cupolillo, E. New Insights on Taxonomy, Phylogeny and Population Genetics of Leishmania (Viannia) Parasites Based on Multilocus Sequence Analysis. PLoS Negl. Trop. Dis. 2012, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Marco, J.D.; Barroso, P.A.; Locatelli, F.M.; Cajal, S.P.; Hoyos, C.L.; Nevot, M.C.; Lauthier, J.J.; Tomasini, N.; Juarez, M.; Estévez, J.O.; et al. Multilocus sequence typing approach for a broader range of species of Leishmania genus: Describing parasite diversity in Argentina. Infect. Genet. Evol. 2015, 30, 308–317. [Google Scholar] [CrossRef]
- Lauthier, J.J.; Ruybal, P.; Barroso, P.A.; Hashiguchi, Y.; Marco, J.D.; Korenaga, M. Development of a Multilocus sequence typing (MLST) scheme for Pan-Leishmania. Acta Trop. 2020, 201, 105189. [Google Scholar] [CrossRef]
- Restrepo, C.M.; Llanes, A.; De La Guardia, C.; Lleonart, R. Genome-wide discovery and development of polymorphic microsatellites from Leishmania panamensis parasites circulating in central Panama. Parasites Vectors 2015, 8, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Valderrama, A.; Gottdenker, N.; Cerezo, L.; Minakawa, N.; Saldaña, A.; Calzada, J.E.; Chaves, L.F. Macroecological patterns of American Cutaneous Leishmaniasis transmission across the health areas of Panamá (1980–2012). Parasite Epidemiol. Control 2016, 1, 42–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Marlow, M.A.; Boité, M.C.; Ferreira, G.E.; Steindel, M.; Cupolillo, E. Multilocus Sequence Analysis for Leishmania braziliensis Outbreak Investigation. PLoS Negl. Trop. Dis. 2014, 8, e2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, G.; Hernández, C.; Ayala, M.S.; Flórez, C.; Teherán, A.A.; Ramírez, J.D. Evaluation of a Multilocus Sequence Typing (MLST) scheme for Leishmania (Viannia) braziliensis and Leishmania (Viannia) panamensis in Colombia. Parasites Vectors 2017, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Tsukayama, P.; Lucas, C.; Bacon, D.J. Typing of four genetic loci discriminates among closely related species of New World Leishmania. Int. J. Parasitol. 2009, 39, 355–362. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Varlamov, A.; Vaskin, Y.; Efremov, I.; German Grehov, O.G.; Kandrov, D.; Rasputin, K.; Syabro, M.; et al. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Rozas, R. DnaSP, DNA sequence polymorphism: An interactive program for estimating population genetics parameters from DNA sequence data. Comput. Appl. Biosci. 1995, 11, 621–625. [Google Scholar] [CrossRef]
- Tomasini, N.; Lauthier, J.J.; Llewellyn, M.S.; Diosque, P. MLSTest: Novel software for multi-locus sequence data analysis in eukaryotic organisms. Infect. Genet. Evol. 2013, 20, 188–196. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. Sea view version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, M.; Sousa, A.; Ramirez, M.; Francisco, A.P.; Carriço, J.A.; Vaz, C. PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 2017, 33, 128–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Veland, N.; Boggild, A.K.; Valencia, C.; Valencia, B.M.; Llanos-Cuentas, A.; Van Der Auwera, G.; Dujardin, J.C.; Arevalo, J. Leishmania (Viannia) species identification on clinical samples from cutaneous leishmaniasis patients in Peru: Assessment of a molecular stepwise approach. J. Clin. Microbiol. 2012, 50, 495–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaría, A.M.; Vásquez, V.; Rigg, C.; Samudio, F.; Moreno, D.; Romero, L.; Saldaña, A.; Chaves, L.F.; Calzada, J.E. Plasmodium vivax genetic diversity in Panama: Challenges for malaria elimination in Mesoamerica. Pathogens 2021, 10, 989. [Google Scholar] [CrossRef]
- Anam, M.; Idiap, E. Instituto Geográfico Nacional “Tommy Guardia” Atlas Nacional de la República de Panamá, 5th ed.; Instituto Tommy Guardia: Panama City, Panama, 2016; ISBN 978-9962-11-048-4. [Google Scholar]
- Chaves, L.F.; Calzada, J.E.; Valderrama, A.; Saldaña, A. Cutaneous Leishmaniasis and Sand Fly Fluctuations Are Associated with El Niño in Panamá. PLoS Negl. Trop. Dis. 2014, 8, e3210. [Google Scholar] [CrossRef] [Green Version]
- Ready, P.D. Leishmaniasis emergence and climate change. OIE Rev. Sci. Tech. 2008, 27, 399–412. [Google Scholar] [CrossRef]
- Hlavacova, J.; Votypka, J.; Volf, P. The effect of temperature on Leishmania (Kinetoplastida: Trypanosomatidae) development in sand flies. J. Med. Entomol. 2013, 50, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Llanes, A.; Cruz, G.; Morán, M.; Vega, C.; Pineda, V.J.; Ríos, M.; Penagos, H.; Suárez, J.A.; Saldaña, A.; Lleonart, R.; et al. Genomic diversity and genetic variation of Leishmania panamensis within its endemic range. Infect. Genet. Evol. 2022, 103, 105342. [Google Scholar] [CrossRef]
- Valderrama, A.; Tavares, M.G.; Filho, J.D.A. Phylogeography of the Lutzomyia gomezi (Diptera: Phlebotominae) on the Panama Isthmus. Parasites Vectors 2014, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rougeron, V.; Bañuls, A.L.; Carme, B.; Simon, S.; Couppié, P.; Nacher, M.; Hide, M.; De Meeûs, T. Reproductive strategies and population structure in Leishmania: Substantial amount of sex in Leishmania Viannia guyanensis. Mol. Ecol. 2011, 20, 3116–3127. [Google Scholar] [CrossRef] [PubMed]
- Rougeron, V.; De Meeûs, T.; Hide, M.; Waleckx, E.; Bermudez, H.; Arevalo, J.; Llanos-Cuentas, A.; Dujardin, J.C.; De Doncker, S.; Le Ray, D.; et al. Extreme inbreeding in Leishmania braziliensis. Proc. Natl. Acad. Sci. USA 2009, 106, 10224–10229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrer, A.; Christensen, H.A. Epidemiological patterns of cutaneous leishmaniasis in Panama. III. Endemic persistence of the disease. Am. J. Trop. Med. Hyg. 1976, 25, 54–58. [Google Scholar] [CrossRef]
- González, K.; Calzada, J.E.; Saldaña, A.; Rigg, C.A.; Alvarado, G.; Rodríguez-Herrera, B.; Kitron, U.D.; Adler, G.H.; Gottdenker, N.L.; Chaves, L.F.; et al. Survey of wild mammal hosts of cutaneous leishmaniasis parasites in Panamá and Costa Rica. Trop. Med. Health 2015, 43, 75–78. [Google Scholar] [CrossRef] [Green Version]
- Dutari, L.C.; Loaiza, J.R. American Cutaneous Leishmaniasis in Panama: A historical review of entomological studies on anthropophilic Lutzomyia sand fly species. Parasites Vectors 2014, 7, 218. [Google Scholar] [CrossRef] [Green Version]
- Christensen, H.A.; De Vasquez, A.M. The tree-buttress biotope: A pathobiocenose of Leishmania braziliensis. Am. J. Trop. Med. Hyg. 1982, 31, 243–251. [Google Scholar] [CrossRef]
- Herrer, A.; Christensen, H.A.; Beumer, R.J. Reservoir hosts of cutaneous leishmaniasis among Panamanian forest mammals. Am. J. Trop. Med. Hyg. 1973, 22, 585–591. [Google Scholar] [CrossRef]
- Valderrama, A.; Tavares, M.G.; Filho, J.D.A. Anthropogenic influence on the distribution, abundance, and diversity of sandfly species (Diptera: Phlebotominae: Psychodidae), vectors of cutaneous leishmaniasis in Panama. Mem. Inst. Oswaldo Cruz 2011, 106, 1024–1031. [Google Scholar] [CrossRef] [Green Version]
- PAHO. Manual de Procedimientos para Vigilancia y Control de Las Leishmaniasis en las Américas; Organización Panamericana de la Salud: Washington, DC, USA, 2019; ISBN 978-92-75-32063-1. [Google Scholar]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.B.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef]
- García-Moreno, J.; Navarro-Sigüenza, A.G.; Peterson, A.T.; Sánchez-González, L.A. Genetic variation coincides with geographic structure in the common bush-tanager (Chlorospingus opthalmicus) complex from Mexico. Mol. Phylogenet. Evol 2004, 206, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Patino, L.H.; Muñoz, M.; Cruz-Saavedra, L.; Muskus, C.; Ramírez, J.D. Genomic Diversification, Structural Plasticity, and Hybridization in Leishmania (Viannia) braziliensis. Front. Cell. Infect. Microbiol. 2020, 10, 582196. [Google Scholar] [CrossRef] [PubMed]
- Patiño, L.H.; Muñoz, M.; Pavia, P.; Muskus, C.; Shaban, M.; Paniz-Mondolfi, A.; Ramírez, J.D. Filling the gaps in Leishmania naiffi and Leishmania guyanensis genome plasticity. G3 2022, 12, jkab377. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Enzyme Entry | Gene Length (bp) | Chromosomal Location | Amplicon Size (bp) | Primers Sequences | Reference |
|---|---|---|---|---|---|---|
| Heat Shock Protein 70 | AAG01344.1 | 2566 | 28 | 1364 | PLeishF: GATGGTGCTGCTGAAGATGA PLeishR: GGTCATGATCGGGTTGCATR | This study |
| Glucose-6-phosphate isomerase | EC 5.3.1.9 | 2084 | 12 | 1745 | GPIextF: AAT GTT CTT CAT ACC CCT CT GPIextR: TTC CGT CCG TCT CCT G GPIintF: TGG GAT TGG CGG CAG CGA CCTT GPIintR: CGC CAC AGG TAC TGG TCG T | [27] |
| Alanine aminotransferase | EC 2.6.1.21 | 1493 | 12 | 589 | ALAT.F: GTGTGCATCAACCCMGGGAA ALAT.R: CGTTCAGCTCCTCGTTCCGC | [20] |
| Aconitase | EC 4.2.1.3 | 2690 | 18 | 579 | ACO.F: CAAGTTCCTGRCGTCTCTGC ACO.R: GAGTCCGGGTATAGCAKCCC | [20] |
| Loci | s | h | hd | π | θ |
|---|---|---|---|---|---|
| HSP70 | 2 | 3 | 0.029 | 0.00002 | 0.00028 |
| GPI | 12 | 6 | 0.099 | 0.00030 | 0.00217 |
| ALAT | 2 | 3 | 0.057 | 0.00012 | 0.00077 |
| Aconitase | 4 | 5 | 0.290 | 0.00067 | 0.00162 |
| Western N = 34 | Eastern N = 35 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Marker | s | h | hd | π | ϴ | s | h | hd | π | ϴ |
| HSP70 | 2 | 3 | 0.060 | 0.00005 | 0.00032 | 0 | 1 | 0 | 0 | 0 |
| GPI | 9 | 4 | 0.118 | 0.00051 | 0.00189 | 3 | 3 | 0.082 | 0.00011 | 0.00062 |
| ALAT | 1 | 2 | 0.060 | 0.00013 | 0.00045 | 1 | 2 | 0.055 | 0.00012 | 0.00044 |
| Aconitase | 3 | 4 | 0.392 | 0.00091 | 0.00140 | 2 | 3 | 0.182 | 0.00041 | 0.00092 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendieta, D.; Vásquez, V.; Jaén, L.; Pineda, V.; Saldaña, A.; Calzada, J.E.; Samudio, F. Insights into the Genetic Diversity of Leishmania (Viannia) panamensis in Panama, Inferred via Multilocus Sequence Typing (MLST). Pathogens 2023, 12, 747. https://doi.org/10.3390/pathogens12050747
Mendieta D, Vásquez V, Jaén L, Pineda V, Saldaña A, Calzada JE, Samudio F. Insights into the Genetic Diversity of Leishmania (Viannia) panamensis in Panama, Inferred via Multilocus Sequence Typing (MLST). Pathogens. 2023; 12(5):747. https://doi.org/10.3390/pathogens12050747
Chicago/Turabian StyleMendieta, Daniel, Vanessa Vásquez, Luis Jaén, Vanessa Pineda, Azael Saldaña, José Eduardo Calzada, and Franklyn Samudio. 2023. "Insights into the Genetic Diversity of Leishmania (Viannia) panamensis in Panama, Inferred via Multilocus Sequence Typing (MLST)" Pathogens 12, no. 5: 747. https://doi.org/10.3390/pathogens12050747
APA StyleMendieta, D., Vásquez, V., Jaén, L., Pineda, V., Saldaña, A., Calzada, J. E., & Samudio, F. (2023). Insights into the Genetic Diversity of Leishmania (Viannia) panamensis in Panama, Inferred via Multilocus Sequence Typing (MLST). Pathogens, 12(5), 747. https://doi.org/10.3390/pathogens12050747