
Deciphering the Role of Protein Phosphatases in Apicomplexa: The Future of Innovative Therapeutics?

 ,
,
Abstract
:1. The Impact of Apicomplexa in Human Health: An Overview
2. Apicomplexan Serine/Threonine and Tyrosine Phosphatome
2.1. Key Differences between the Apicomplexan and Human Phosphatome
2.2. General Characteristics of the Apicomplexan Serine/Threonine Phosphatome
2.2.1. PPPL Superfamily
2.2.2. PPM (Protein Phosphatase Mn2+ or Mg2+-Dependent or PP2C) Superfamily
2.2.3. HAD Superfamily
2.3. General Characteristics of the Apicomplexan Tyrosine Phosphatome
2.3.1. CC1 Superfamily
2.3.2. CC2 and CC3 Superfamilies
2.3.3. Other Tyrosine Phosphatases
3. The Function of Protein Phosphatases in Apicomplexa: Where Do We Stand?
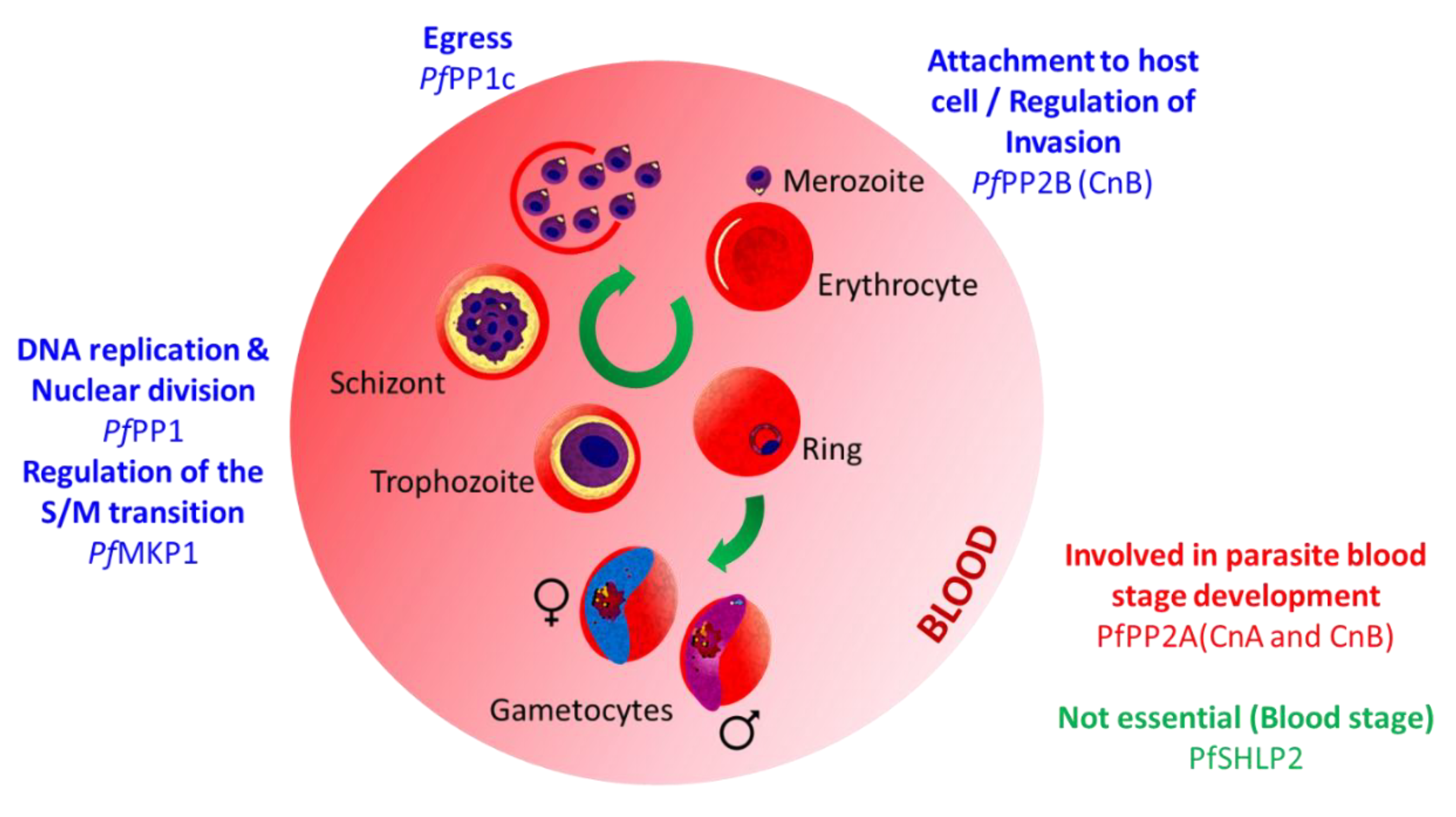
3.1. Functional Characterization of Plasmodium Proteins Phosphatases
3.1.1. PPPL Fold
- Phosphoprotein Phosphatases (PPP) family
Protein Phosphatase Type 1 (PP1)
Calcineurin (PP2B; CnA, CnB)
PP5
Protein Phosphatase Containing Kelch-like Domains (PPKL)
Schewanella-like Phosphatases
- 2.
- Purple acid phosphatase (PAP) family
GAP50/SAP (Secreted Acid Phosphatase)
UIS2
3.1.2. Protein Phosphatase Mn2+ or Mg2+ Dependent (PPM) Fold
- 3.
- Protein Phosphatases Mn2+ or Mg2+ dependent (PPM) family
- 4.
- PfMKP1
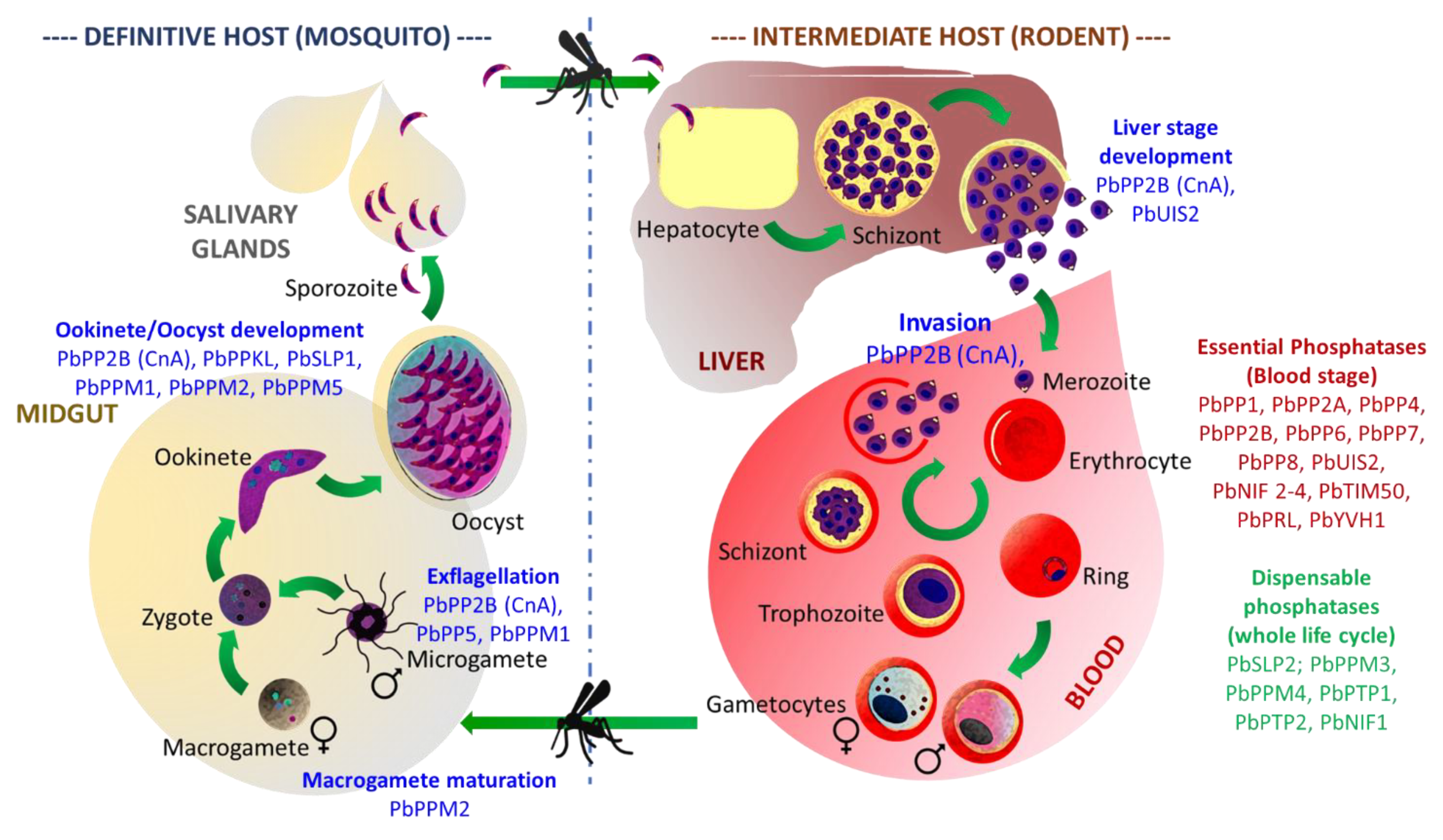
3.2. Functional Characterization of Toxoplasma gondii Proteins Phosphatases
3.2.1. PPPL Fold
- 5.
- Phosphoprotein Phosphatases (PPP) family
Protein Phosphatase Type 1 (PP1)
PP2A and PP2B (Calcineurin)
- 6.
- Purple acid Phosphatase (PAP) family
GAP50/SAP (Secreted Acid Phosphatase)
GRA44 (TGGT1_228170 Secreted Acid Phosphatase)
3.2.2. Protein Phosphatase Mn2+ or Mg2+ Dependent (PPM) Fold
- 7.
- Protein Phosphatases Mn2+ or Mg2+ dependent (PPM) family
- 8.
- Aspartate-based phosphatase family (FCP/SCP)
4. Therapeutic Potential of Protein Phosphatases as Drug Targets: The Case of Plasmodium
4.1. Targeting Phosphatases to Treat Human Diseases: Two Decisive Cases
4.2. Is Targeting Phosphatases the Future of Anti-Plasmodium Therapeutics?
4.3. Targeting the PPP Family
4.3.1. PP1 and PP2A
4.3.2. Calcineurin (PP2B)
4.3.3. PP4 and PP6 In Silico Analysis of Druggability
4.4. Targeting the Tyrosine Phosphatases
4.4.1. In Silico Docking Analysis of PRL
4.4.2. In Silico Docking Analysis of PfMKP1
5. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adl, S.M.; Leander, B.S.; Simpson, A.G.B.; Archibald, J.M.; Anderson, O.R.; Bass, D.; Bowser, S.S.; Brugerolle, G.; Farmer, M.A.; Karpov, S.; et al. Diversity, Nomenclature, and Taxonomy of Protists. Syst. Biol. 2007, 56, 684–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackman, M.J.; Bannister, L.H. Apical organelles of Apicomplexa: Biology and isolation by subcellular fractionation. Mol. Biochem. Parasitol. 2001, 117, 11–25. [Google Scholar] [CrossRef]
- Fichera, M.E.; Roos, D.S. A plastid organelle as a drug target in apicomplexan parasites. Nature 1997, 390, 407–409. [Google Scholar] [CrossRef]
- McFadden, G.I. Apicoplast. Curr. Biol. 2014, 24, R262–R263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomaki, E.D.; Kolisko, M. There Is Treasure Everywhere: Reductive Plastid Evolution in Apicomplexa in Light of Their Close Relatives. Biomolecules 2019, 9, 378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health, O. World Malaria Report 2020: 20 Years of Global Progress and Challenges; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Hajj, R.E.; Tawk, L.; Itani, S.; Hamie, M.; Ezzeddine, J.; El Sabban, M.; El Hajj, H. Toxoplasmosis: Current and Emerging Parasite Druggable Targets. Microorganisms 2021, 9, 2531. [Google Scholar] [CrossRef]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Djurković-Djaković, O.; Dupouy-Camet, J.; Van der Giessen, J.; Dubey, J.P. Toxoplasmosis: Overview from a One Health perspective. Food Waterborne Parasitol. 2019, 15, e00054. [Google Scholar] [CrossRef]
- Kezai, A.M.; Lecoeur, C.; Hot, D.; Bounechada, M.; Alouani, M.L.; Marion, S. Association between schizophrenia and Toxoplasma gondii infection in Algeria. Psychiat Res. 2020, 291, 113293. [Google Scholar] [CrossRef]
- Fuglewicz, A.J.; Piotrowski, P.; Stodolak, A. Relationship between toxoplasmosis and schizophrenia: A review. Adv. Clin. Exp. Med. 2017, 26, 1031–1036. [Google Scholar] [CrossRef]
- Fabiani, S.; Pinto, B.; Bonuccelli, U.; Bruschi, F. Neurobiological studies on the relationship between toxoplasmosis and neuropsychiatric diseases. J. Neurol. Sci 2015, 351, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngoungou, E.B.; Bhalla, D.; Nzoghe, A.; Dardé, M.-L.; Preux, P.-M. Toxoplasmosis and epilepsy--systematic review and meta analysis. PLoS Negl. Trop. Dis. 2015, 9, e0003525. [Google Scholar] [CrossRef]
- Vannier, E.; Krause, P.J. Human Babesiosis. N. Eng. J. Med. 2012, 366, 2397–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Ryan, U.M.; Xiao, L. Genetic Diversity and Population Structure of Cryptosporidium. Trends Parasitol 2018, 34, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Platts-Mills, J.A.; Babji, S.; Bodhidatta, L.; Gratz, J.; Haque, R.; Havt, A.; McCormick, B.J.; McGrath, M.; Olortegui, M.P.; Samie, A.; et al. Pathogen-specific burdens of community diarrhoea in developing countries: A multisite birth cohort study (MAL-ED). Lancet Glob. Health 2015, 3, e564–e575. [Google Scholar] [CrossRef] [Green Version]
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F.; et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef]
- O’Connor, R.M.; Shaffie, R.; Kang, G.; Ward, H.D. Cryptosporidiosis in patients with HIV/AIDS. AIDS 2011, 25, 549–560. [Google Scholar] [CrossRef]
- Sawant, M.; Baydoun, M.; Creusy, C.; Chabé, M.; Viscogliosi, E.; Certad, G.; Benamrouz-Vanneste, S. Cryptosporidium and Colon Cancer: Cause or Consequence? Microorganisms 2020, 8, 1665. [Google Scholar] [CrossRef]
- Van Dooren, G.G.; Striepen, B. The Algal Past and Parasite Present of the Apicoplast. Annu. Rev. Microbiol. 2013, 67, 271–289. [Google Scholar] [CrossRef]
- Gubbels, M.-J.; Coppens, I.; Zarringhalam, K.; Duraisingh, M.T.; Engelberg, K. The Modular Circuitry of Apicomplexan Cell Division Plasticity. Front. Cell Infect. Microbiol. 2021, 11, 670049. [Google Scholar] [CrossRef]
- Moorhead, G.B.G.; De Wever, V.; Templeton, G.; Kerk, D. Evolution of protein phosphatases in plants and animals. Biochem. J. 2009, 417, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.J.; Dixon, J.E.; Manning, G. Genomics and evolution of protein phosphatases. Sci. Signal. 2017, 10, eaag1796. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Saavedra, D.; Gabaldón, T.; Barton, G.J.; Langsley, G.; Doerig, C. The kinomes of apicomplexan parasites. Microbes Infect. 2012, 14, 796–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Saavedra, D.; Barton, G.J. Classification and functional annotation of eukaryotic protein kinases. Proteins 2007, 68, 893–914. [Google Scholar] [CrossRef]
- Yang, C.; Arrizabalaga, G. The serine/threonine phosphatases of apicomplexan parasites. Mol. Microbiol. 2017, 106, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Wilkes, J.M.; Doerig, C. The protein-phosphatome of the human malaria parasite Plasmodium falciparum. BMC Genom. 2008, 9, 412. [Google Scholar] [CrossRef] [Green Version]
- Phosphatase Wiki—Protein Phosphatase Classification and Evolution. Available online: http://phosphatome.net/wiki (accessed on 1 September 2021).
- Treeck, M.; Sanders, J.L.; Elias, J.E.; Boothroyd, J.C. The phosphoproteomes of Plasmodium falciparum and Toxoplasma gondii reveal unusual adaptations within and beyond the parasites’ boundaries. Cell Host Microbe 2011, 10, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Hunter, T. The genesis of tyrosine phosphorylation. Cold Spring Harb. Perspect. Biol. 2014, 6, a020644. [Google Scholar] [CrossRef] [Green Version]
- Spalinger, M.R.; Lang, S.; Gottier, C.; Dai, X.; Rawlings, D.J.; Chan, A.C.; Rogler, G.; Scharl, M. PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy 2017, 13, 1590–1601. [Google Scholar] [CrossRef] [Green Version]
- Penafuerte, C.; Perez-Quintero, L.A.; Vinette, V.; Hatzihristidis, T.; Tremblay, M.L. Mining the Complex Family of Protein Tyrosine Phosphatases for Checkpoint Regulators in Immunity. In Emerging Concepts Targeting Immune Checkpoints in Cancer and Autoimmunity; Yoshimura, A., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 191–214. [Google Scholar]
- Wade, F.; Belhaj, K.; Poizat, C. Protein tyrosine phosphatases in cardiac physiology and pathophysiology. Heart Fail. Rev. 2018, 23, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.-P.; Qi, X.-W.; Sun, N.; Sun, Y.-Y.; Zhang, Y.; Tan, X.-N.; Ding, J.; Han, F.; Zhang, Y. The emerging roles of dual-specificity phosphatases and their specific characteristics in human cancer. Biochim. Biophys. Acta 2021, 1876, 188562. [Google Scholar] [CrossRef] [PubMed]
- Ruckert, M.T.; de Andrade, P.V.; Santos, V.S.; Silveira, V.S. Protein tyrosine phosphatases: Promising targets in pancreatic ductal adenocarcinoma. Cell. Mol. Life Sci. CMLS 2019, 76, 2571–2592. [Google Scholar] [CrossRef] [PubMed]
- Frankson, R.; Yu, Z.-H.; Bai, Y.; Li, Q.; Zhang, R.-Y.; Zhang, Z.-Y. Therapeutic Targeting of Oncogenic Tyrosine Phosphatases. Cancer Res. 2017, 77, 5701–5705. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Pendyala, P.R.; Ayong, L.; Eatrides, J.; Schreiber, M.; Pham, C.; Chakrabarti, R.; Fidock, D.A.; Allen, C.M.; Chakrabarti, D. Characterization of a PRL protein tyrosine phosphatase from Plasmodium falciparum. Mol. Biochem. Parasitol. 2008, 158, 1–10. [Google Scholar] [CrossRef]
- Kumar, R.; Musiyenko, A.; Cioffi, E.; Oldenburg, A.; Adams, B.; Bitko, V.; Krishna, S.S.; Barik, S. A zinc-binding dual-specificity YVH1 phosphatase in the malaria parasite, Plasm.modium falciparum, and its interaction with the nuclear protein, pescadillo. Mol. Biochem. Parasitol. 2004, 133, 297–310. [Google Scholar] [CrossRef]
- Balu, B.; Campbell, C.; Sedillo, J.; Maher, S.; Singh, N.; Thomas, P.; Zhang, M.; Pance, A.; Otto, T.D.; Rayner, J.C.; et al. Atypical mitogen-activated protein kinase phosphatase implicated in regulating transition from pre-S-Phase asexual intraerythrocytic development of Plasmodium falciparum. Eukaryot. Cell 2013, 12, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Guttery, D.S.; Pandey, R.; Ferguson, D.J.; Wall, R.J.; Brady, D.; Gupta, D.; Holder, A.A.; Tewari, R. Plasmodium DEH is ER-localized and crucial for oocyst mitotic division during malaria transmission. Life Sci. Alliance 2020, 3, e202000879. [Google Scholar] [CrossRef]
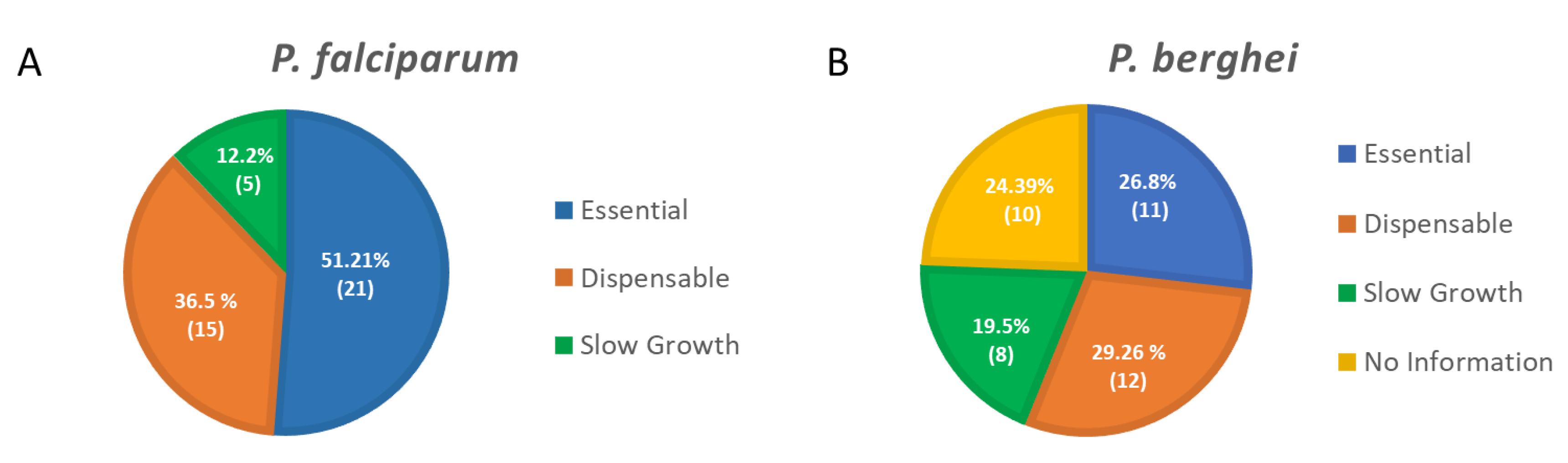
- Zhang, M.; Wang, C.; Otto, T.D.; Oberstaller, J.; Liao, X.; Adapa, S.R.; Udenze, K.; Bronner, I.F.; Casandra, D.; Mayho, M.; et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 2018, 360, eaap7847. [Google Scholar] [CrossRef] [Green Version]
- Bushell, E.; Gomes, A.R.; Sanderson, T.; Anar, B.; Girling, G.; Herd, C.; Metcalf, T.; Modrzynska, K.; Schwach, F.; Martin, R.E.; et al. Functional Profiling of a Plasmodium Genome Reveals an Abundance of Essential Genes. Cell 2017, 170, 260–272.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidik, S.M.; Huet, D.; Ganesan, S.M.; Huynh, M.H.; Wang, T.; Nasamu, A.S.; Thiru, P.; Saeij, J.P.J.; Carruthers, V.B.; Niles, J.C.; et al. A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 2016, 166, 1423–1435.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceulemans, H.; Bollen, M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol. Rev. 2004, 84, 1–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbinnen, I.; Ferreira, M.; Bollen, M. Biogenesis and activity regulation of protein phosphatase 1. Biochem. Soc. Trans. 2017, 45, 89–99. [Google Scholar] [CrossRef]
- Daher, W.; Browaeys, E.; Pierrot, C.; Jouin, H.; Dive, D.; Meurice, E.; Dissous, C.; Capron, M.; Tomavo, S.; Doerig, C.; et al. Regulation of protein phosphatase type 1 and cell cycle progression by PfLRR1, a novel leucine-rich repeat protein of the human malaria parasite Plasmodium falciparum. Mol. Microbiol. 2006, 60, 578–590. [Google Scholar] [CrossRef]
- Hollin, T.; De Witte, C.; Freville, A.; Guerrera, I.C.; Chhuon, C.; Saliou, J.M.; Herbert, F.; Pierrot, C.; Khalife, J. Essential role of GEXP15, a specific Protein Phosphatase type 1 partner, in Plasmodium berghei in asexual erythrocytic proliferation and transmission. PLoS Pathog. 2019, 15, e1007973. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.S.; Miliu, A.; Paulo, J.A.; Goldberg, J.M.; Bonilla, A.M.; Berry, L.; Séveno, M.; Braun-Breton, C.; Kosber, A.L.; Elsworth, B.; et al. Co-option of Plasmodium falciparum PP1 for egress from host erythrocytes. Nat. Commun. 2020, 11, 3532. [Google Scholar] [CrossRef]
- Blisnick, T.; Vincensini, L.; Fall, G.; Braun-Breton, C. Protein phosphatase 1, a Plasmodium falciparum essential enzyme, is exported to the host cell and implicated in the release of infectious merozoites. Cell. Microbiol. 2006, 8, 591–601. [Google Scholar] [CrossRef]
- Bhattacharyya, M.K.; Hong, Z.; Kongkasuriyachai, D.; Kumar, N. Plasmodium falciparum protein phosphatase type 1 functionally complements a glc7 mutant in Saccharomyces cerevisiae. Int. J. Parasitol. 2002, 32, 739–747. [Google Scholar] [CrossRef]
- Zeeshan, M.; Pandey, R.; Subudhi, A.K.; Ferguson, D.J.P.; Kaur, G.; Rashpa, R.; Nugmanova, R.; Brady, D.; Bottrill, A.R.; Vaughan, S.; et al. Protein phosphatase 1 regulates atypical mitotic and meiotic division in Plasmodium sexual stages. Commun. Biol. 2021, 4, 760. [Google Scholar] [CrossRef]
- Combe, A.; Giovannini, D.; Carvalho, T.G.; Spath, S.; Boisson, B.; Loussert, C.; Thiberge, S.; Lacroix, C.; Gueirard, P.; Ménard, R. Clonal Conditional Mutagenesis in Malaria Parasites. Cell Host Microbe 2009, 5, 386–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollin, T.; De Witte, C.; Lenne, A.; Pierrot, C.; Khalife, J. Analysis of the interactome of the Ser/Thr Protein Phosphatase type 1 in Plasmodium falciparum. BMC Genom. 2016, 17, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freville, A.; Cailliau-Maggio, K.; Pierrot, C.; Tellier, G.; Kalamou, H.; Lafitte, S.; Martoriati, A.; Pierce, R.J.; Bodart, J.F.; Khalife, J. Plasmodium falciparum encodes a conserved active inhibitor-2 for Protein Phosphatase type 1: Perspectives for novel anti-plasmodial therapy. BMC Biol. 2013, 11, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freville, A.; Landrieu, I.; Garcia-Gimeno, M.A.; Vicogne, J.; Montbarbon, M.; Bertin, B.; Verger, A.; Kalamou, H.; Sanz, P.; Werkmeister, E.; et al. Plasmodium falciparum inhib.bitor-3 homolog increases protein phosphatase type 1 activity and is essential for parasitic survival. J. Biol. Chem. 2012, 287, 1306–1321. [Google Scholar] [CrossRef] [Green Version]
- Freville, A.; Tellier, G.; Vandomme, A.; Pierrot, C.; Vicogne, J.; Cantrelle, F.X.; Martoriati, A.; Cailliau-Maggio, K.; Khalife, J.; Landrieu, I. Identification of a Plasmodium falciparum inhibitor-2 motif involved in the binding and regulation activity of protein phosphatase type 1. FEBS J. 2014, 281, 4519–4534. [Google Scholar] [CrossRef]
- Gnangnon, B.; Fréville, A.; Cailliau, K.; Leroy, C.; De Witte, C.; Tulasne, D.; Martoriarti, A.; Jung, V.; Guerrera, I.C.; Marion, S.; et al. Plasmodium pseudo-Tyrosine Kinase-like binds PP1 and SERA5 and is exported to host erythrocytes. Sci. Rep. 2019, 9, 8120. [Google Scholar] [CrossRef] [Green Version]
- Lenne, A.; De Witte, C.; Tellier, G.; Hollin, T.; Aliouat, E.M.; Martoriati, A.; Cailliau, K.; Saliou, J.-M.; Khalife, J.; Pierrot, C. Characterization of a Protein Phosphatase Type-1 and a Kinase Anchoring Protein in Plasmodium falciparum. Front. Microbiol. 2018, 9, 2617. [Google Scholar] [CrossRef]
- Pierrot, C.; Zhang, X.; Zhangi, G.; Freville, A.; Rebollo, A.; Khalife, J. Peptides derived from Plasmodium falciparum leucine-rich repeat 1 bind to serine/threonine phosphatase type 1 and inhibit parasite growth in vitro. Drug Des. Dev. Ther. 2018, 12, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Tellier, G.; Lenne, A.; Cailliau-Maggio, K.; Cabezas-Cruz, A.; Valdes, J.J.; Martoriati, A.; Aliouat el, M.; Gosset, P.; Delaire, B.; Freville, A.; et al. Identification of Plasmodium falciparum Translation Initiation eIF2beta Subunit: Direct Interaction with Protein Phosphatase Type 1. Front. Microbiol. 2016, 7, 777. [Google Scholar] [CrossRef]
- Khalife, J.; Fréville, A.; Gnangnon, B.; Pierrot, C. The Multifaceted Role of Protein Phosphatase 1 in Plasmodium. Trends Parasitol. 2021, 37, 154–164. [Google Scholar] [CrossRef]
- Rusnak, F.; Mertz, P. Calcineurin: Form and Function. Physiol. Rev. 2000, 80, 1483–1521. [Google Scholar] [CrossRef] [PubMed]
- Klee, C.B.; Ren, H.; Wang, X. Regulation of the Calmodulin-stimulated Protein Phosphatase, Calcineurin. J. Biol. Chem. 1998, 273, 13367–13370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, S.; May, T.; Berriman, M.; Del Vecchio, C.; Fairlamb, A.H.; Chakrabarti, D.; Barik, S. Characterization of protein Ser/Thr phosphatases of the malaria parasite, Plasmodium falciparum: Inhibition of the parasitic calcineurin by cyclophilin-cyclosporin complex. Mol. Biochem. Parasitol. 1999, 99, 167–181. [Google Scholar] [CrossRef]
- Liu, J.; Farmer, J.D.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Singh, S.; More, K.R.; Chitnis, C.E. Role of calcineurin and actin dynamics in regulated secretion of microneme proteins in Plasmodium falciparum merozoites during erythrocyte invasion. Cell. Microbiol. 2014, 16, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.S.; Saha, S.; Engelberg, K.; Jiang, R.H.Y.; Coleman, B.I.; Kosber, A.L.; Chen, C.-T.; Ganter, M.; Espy, N.; Gilberger, T.W.; et al. Parasite Calcineurin Regulates Host Cell Recognition and Attachment by Apicomplexans. Cell Host Microbe 2015, 18, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, N.; Waters, A.P. Conditional Degradation of Plasmodium Calcineurin Reveals Functions in Parasite Colonization of both Host and Vector. Cell Host Microbe 2015, 18, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Chinkers, M. Targeting of a distinctive protein-serine phosphatase to the protein kinase-like domain of the atrial natriuretic peptide receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 11075–11079. [Google Scholar] [CrossRef] [Green Version]
- Becker, W.; Kentrup, H.; Klumpp, S.; Schultz, J.E.; Joost, H.G. Molecular cloning of a protein serine/threonine phosphatase containing a putative regulatory tetratricopeptide repeat domain. J. Biol. Chem. 1994, 269, 22586–22592. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Sánchez, E.R. Protein phosphatase 5. Int. J. Biochem. Cell Biol. 2008, 40, 2358–2362. [Google Scholar] [CrossRef]
- Dobson, S.; Kar, B.; Kumar, R.; Adams, B.; Barik, S. A novel tetratricopeptide repeat (TPR) containing PP5 serine/threonine protein phosphatase in the malaria parasite, Plasmodium falciparum. BMC Microbiol. 2001, 1, 31. [Google Scholar] [CrossRef] [PubMed]
- Lindenthal, C.; Klinkert, M.Q. Identification and biochemical characterisation of a protein phosphatase 5 homologue from Plasmodium falciparum. Mol. Biochem. Parasitol 2002, 120, 257–268. [Google Scholar] [CrossRef]
- Guttery, D.S.; Poulin, B.; Ramaprasad, A.; Wall, R.J.; Ferguson, D.J.; Brady, D.; Patzewitz, E.M.; Whipple, S.; Straschil, U.; Wright, M.H.; et al. Genome-wide functional analysis of Plasmodium protein phosphatases reveals key regulators of parasite development and differentiation. Cell Host Microbe 2014, 16, 128–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Sun, L.; He, Y.; Wei, H.; Hong, M.; Liu, F.; Liu, Q.; Cao, Y.; Cui, L. Plasmodium berghei serine/threonine protein phosphatase PP5 plays a critical role in male gamete fertility. Int. J. Parasitol Drug 2019, 49, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Kutuzov, M.A.; Andreeva, A.V. Protein Ser/Thr phosphatases with kelch-like repeat domains. Cell. Signal. 2002, 14, 745–750. [Google Scholar] [CrossRef]
- Li, J.L.; Baker, D.A. A putative protein serine/threonine phosphatase from Plasmodium falciparum contains a large N-terminal extension and five unique inserts in the catalytic domain. Mol. Biochem. Parasitol. 1998, 95, 287–295. [Google Scholar] [CrossRef]
- Philip, N.; Vaikkinen, H.J.; Tetley, L.; Waters, A.P. A unique Kelch domain phosphatase in Plasmodium regulates ookinete morphology, motility and invasion. PLoS ONE 2012, 7, e44617. [Google Scholar] [CrossRef] [Green Version]
- Guttery, D.S.; Poulin, B.; Ferguson, D.J.; Szoor, B.; Wickstead, B.; Carroll, P.L.; Ramakrishnan, C.; Brady, D.; Patzewitz, E.M.; Straschil, U.; et al. A unique protein phosphatase with kelch-like domains (PPKL) in Plasmodium modulates ookinete differentiation, motility and invasion. PLoS Pathog. 2012, 8, e1002948. [Google Scholar] [CrossRef]
- Andreeva, A.V.; Kutuzov, M.A. Widespread presence of “bacterial-like” PPP phosphatases in eukaryotes. BMC Evol. Biol. 2004, 4, 47. [Google Scholar] [CrossRef] [Green Version]
- Patzewitz, E.M.; Guttery, D.S.; Poulin, B.; Ramakrishnan, C.; Ferguson, D.J.; Wall, R.J.; Brady, D.; Holder, A.A.; Szoor, B.; Tewari, R. An ancient protein phosphatase, SHLP1, is critical to microneme development in Plasmodium ookinetes and parasite transmission. Cell Rep. 2013, 3, 622–629. [Google Scholar] [CrossRef] [Green Version]
- Miliu, A.; Lebrun, M.; Braun-Breton, C.; Lamarque, M.H. Shelph2, a bacterial-like phosphatase of the malaria parasite Plasmodium falciparum, is dispensable during asexual blood stage. PLoS ONE 2017, 12, e0187073. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pol, S.; Slouka, Z.; Bhattacharjee, S.; Fedotova, Y.; Freed, S.; An, X.; Holder, A.A.; Campanella, E.; Low, P.S.; Mohandas, N.; et al. A bacterial phosphatase-like enzyme of the malaria parasite Plasmodium falciparum possesses tyrosine phosphatase activity and is implicated in the regulation of band 3 dynamics during parasite invasion. Eukaryot. Cell 2013, 12, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldati-Favre, D. Molecular dissection of host cell invasion by the Apicomplexans: The glideosome. Parasite 2008, 15, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baum, J.; Richard, D.; Healer, J.; Rug, M.; Krnajski, Z.; Gilberger, T.W.; Green, J.L.; Holder, A.A.; Cowman, A.F. A conserved molecular motor drives cell invasion and gliding motility across malaria life cycle stages and other apicomplexan parasites. J. Biol. Chem. 2006, 281, 5197–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeoman, J.A.; Hanssen, E.; Maier, A.G.; Klonis, N.; Maco, B.; Baum, J.; Turnbull, L.; Whitchurch, C.B.; Dixon, M.W.; Tilley, L. Tracking Glideosome-associated protein 50 reveals the development and organization of the inner membrane complex of Plasmodium falciparum. Eukaryot. Cell 2011, 10, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dearnley, M.K.; Yeoman, J.A.; Hanssen, E.; Kenny, S.; Turnbull, L.; Whitchurch, C.B.; Tilley, L.; Dixon, M.W.A. Origin, composition, organization and function of the inner membrane complex of Plasmodium falciparum gametocytes. J. Cell Sci. 2012, 125, 2053–2063. [Google Scholar] [CrossRef] [Green Version]
- Simon, N.; Lasonder, E.; Scheuermayer, M.; Kuehn, A.; Tews, S.; Fischer, R.; Zipfel, P.F.; Skerka, C.; Pradel, G. Malaria Parasites Co-opt Human Factor H to Prevent Complement-Mediated Lysis in the Mosquito Midgut. Cell Host Microbe 2013, 13, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Bosch, J.; Paige, M.H.; Vaidya, A.B.; Bergman, L.W.; Hol, W.G. Crystal structure of GAP50, the anchor of the invasion machinery in the inner membrane complex of Plasmodium falciparum. J. Struct. Biol. 2012, 178, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Muller, I.B.; Knockel, J.; Eschbach, M.L.; Bergmann, B.; Walter, R.D.; Wrenger, C. Secretion of an acid phosphatase provides a possible mechanism to acquire host nutrients by Plasmodium falciparum. Cell. Microbiol. 2010, 12, 677–691. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Scheuner, D.; Chen, J.-J.; Kaufman, R.J.; Ron, D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc. Natl. Acad. Sci. USA 2009, 106, 1832–1837. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Mishra, S.; Sakthivel, R.; Fontoura, B.M.; Nussenzweig, V. UIS2: A Unique Phosphatase Required for the Development of Plasmodium Liver Stages. PLoS Pathog. 2016, 12, e1005370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamoun, C.B.; Sullivan, D.J., Jr.; Banerjee, R.; Goldberg, D.E. Identification and characterization of an unusual double serine/threonine protein phosphatase 2C in the malaria parasite Plasmodium falciparum. J. Biol. Chem. 1998, 273, 11241–11247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamoun, C.B.; Goldberg, D.E. Plasmodium protein phosphatase 2C dephosphorylates translation elongation factor 1beta and inhibits its PKC-mediated nucleotide exchange activity in vitro. Mol. Microbiol. 2001, 39, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, K.; Nishida, E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta 2007, 1773, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooq, A.; Chaturvedi, G.; Mujtaba, S.; Plotnikova, O.; Zeng, L.; Dhalluin, C.; Ashton, R.; Zhou, M.-M. Solution Structure of ERK2 Binding Domain of MAPK Phosphatase MKP-3: Structural Insights into MKP-3 Activation by ERK2. Mol. Cell 2001, 7, 387–399. [Google Scholar] [CrossRef]
- Theodosiou, A.; Ashworth, A. MAP kinase phosphatases. Genome Biol. 2002, 3, 3009.3001–3009.3010. [Google Scholar] [CrossRef] [Green Version]
- Farooq, A.; Zhou, M.-M. Structure and regulation of MAPK phosphatases. Cell. Signal. 2004, 16, 769–779. [Google Scholar] [CrossRef]
- Delorme, V.; Garcia, A.; Cayla, X.; Tardieux, I. A role for Toxoplasma gondii type 1 ser/thr protein phosphatase in host cell invasion. Microbes Infect. 2002, 4, 271–278. [Google Scholar] [CrossRef]
- Daher, W.; Oria, G.; Fauquenoy, S.; Cailliau, K.; Browaeys, E.; Tomavo, S.; Khalife, J. A Toxoplasma gondii leucine-rich repeat protein binds phosphatase type 1 protein and negatively regulates its activity. Eukaryot. Cell 2007, 6, 1606–1617. [Google Scholar] [CrossRef] [Green Version]
- Deveuve, Q.; Lesage, K.; Mouveaux, T.; Gissot, M. The Toxoplasma gondii inhibitor-2 regulates protein phosphatase 1 activity through multiple motifs. Parasitol. Res. 2017, 116, 2417–2426. [Google Scholar] [CrossRef]
- Jiang, Y. Regulation of the cell cycle by protein phosphatase 2A in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2006, 70, 440–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, C.R.; Egarter, S.; Gow, M.; Jiménez-Ruiz, E.; Ferguson, D.J.P.; Meissner, M. Gliding Associated Proteins Play Essential Roles during the Formation of the Inner Membrane Complex of Toxoplasma gondii. PLoS Pathog. 2016, 12, e1005403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakely, W.J.; Holmes, M.J.; Arrizabalaga, G. The Secreted Acid Phosphatase Domain-Containing GRA44 from Toxoplasma gondii Is Required for c-Myc Induction in Infected Cells. mSphere 2020, 5, e00877-00819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Broncel, M.; Dominicus, C.; Sampson, E.; Blakely, W.J.; Treeck, M.; Arrizabalaga, G. A plasma membrane localized protein phosphatase in Toxoplasma gondii, PPM5C, regulates attachment to host cells. Sci. Rep. 2019, 9, 5924. [Google Scholar] [CrossRef] [Green Version]
- Cygan, A.M.; Theisen, T.C.; Mendoza, A.G.; Marino, N.D.; Panas, M.W.; Boothroyd, J.C. Coimmunoprecipitation with MYR1 Identifies Three Additional Proteins within the Toxoplasma gondii Parasitophorous Vacuole Required for Translocation of Dense Granule Effectors into Host Cells. mSphere 2020, 5, e00858-00819. [Google Scholar] [CrossRef] [Green Version]
- Mayoral, J.; Tomita, T.; Tu, V.; Aguilan, J.T.; Sidoli, S.; Weiss, L.M. Toxoplasma gondii PPM3C, a secreted protein phosphatase, affects parasitophorous vacuole effector export. PLoS Pathog. 2020, 16, e1008771. [Google Scholar] [CrossRef]
- Jan, G.; Delorme, V.; Saksouk, N.; Abrivard, M.; Gonzalez, V.; Cayla, X.; Hakimi, M.-A.; Tardieux, I. A Toxoplasma type 2C serine-threonine phosphatase is involved in parasite growth in the mammalian host cell. Microbes Infect. 2009, 11, 935–945. [Google Scholar] [CrossRef]
- Delorme, V.; Cayla, X.; Faure, G.; Garcia, A.; Tardieux, I. Actin dynamics is controlled by a casein kinase II and phosphatase 2C interplay on Toxoplasma gondii Toxofilin. Mol. Biol. Cell 2003, 14, 1900–1912. [Google Scholar] [CrossRef] [Green Version]
- Delorme-Walker, V.; Abrivard, M.; Lagal, V.; Anderson, K.; Perazzi, A.; Gonzalez, V.; Page, C.; Chauvet, J.; Ochoa, W.; Volkmann, N.; et al. Toxofilin upregulates the host cortical actin cytoskeleton dynamics, facilitating Toxoplasma invasion. J. Cell Sci. 2012, 125, 4333–4342. [Google Scholar] [CrossRef] [Green Version]
- Jan, G.; Delorme, V.; David, V.; Revenu, C.; Rebollo, A.; Cayla, X.; Tardieux, I. The toxofilin-actin-PP2C complex of Toxoplasma: Identification of interacting domains. Biochem. J. 2007, 401, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Ravindran, S.; Turetzky, J.M.; Boothroyd, J.C.; Bradley, P.J. Toxoplasma gondii targets a protein phosphatase 2C to the nuclei of infected host cells. Eukaryot. Cell 2007, 6, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, P.X.; Yao, S.H.; Yao, Y.; Zhou, J.; Li, Q.F.; Cao, Y.H.; He, S.Y. Epitope Analysis and Efficacy Evaluation of Phosphatase 2C (PP2C) DNA Vaccine Against Toxoplasma gondii Infection. J. Parasitol. 2020, 106, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Stiller, J.W.; Cook, M.S. Functional unit of the RNA polymerase II C-terminal domain lies within heptapeptide pairs. Eukaryot. Cell 2004, 3, 735–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braconi Quintaje, S.; Orchard, S. The annotation of both human and mouse kinomes in UniProtKB/Swiss-Prot: One small step in manual annotation, one giant leap for full comprehension of genomes. Mol. Cell. Proteom. MCP 2008, 7, 1409–1419. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Yogesha, S.D.; Mayfield, J.E.; Gill, G.N.; Zhang, Y. Viewing serine/threonine protein phosphatases through the eyes of drug designers. FEBS J. 2013, 280, 4739–4760. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, W.J.A.J.; Elson, A.; Harroch, S.; Pulido, R.; Stoker, A.; den Hertog, J. Protein tyrosine phosphatases in health and disease. FEBS J. 2013, 280, 708–730. [Google Scholar] [CrossRef]
- Khalife, J.; Pierrot, C. Phosphatases are emerging as novel druggable targets in Plasmodium. Future Microbiol. 2016, 11, 603–606. [Google Scholar] [CrossRef] [Green Version]
- Köhn, M. Turn and Face the Strange: A New View on Phosphatases. ACS Cent. Sci. 2020, 6, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-N.P.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Krzyzosiak, A.; Sigurdardottir, A.; Luh, L.; Carrara, M.; Das, I.; Schneider, K.; Bertolotti, A. Target-Based Discovery of an Inhibitor of the Regulatory Phosphatase PPP1R15B. Cell 2018, 174, 1216–1228.e19. [Google Scholar] [CrossRef] [Green Version]
- Anavo Therapeutics. Available online: https://www.anavotx.com/ (accessed on 1 September 2021).
- Pease, B.N.; Huttlin, E.L.; Jedrychowski, M.P.; Talevich, E.; Harmon, J.; Dillman, T.; Kannan, N.; Doerig, C.; Chakrabarti, R.; Gygi, S.P.; et al. Global analysis of protein expression and phosphorylation of three stages of Plasmodium falciparum intraerythrocytic development. J. Proteome Res. 2013, 12, 4028–4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fardilha, M.; Esteves, S.L.; Korrodi-Gregorio, L.; da Cruz e Silva, O.A.; da Cruz e Silva, F.F. The physiological relevance of protein phosphatase 1 and its interacting proteins to health and disease. Curr. Med. Chem. 2010, 17, 3996–4017. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Serine/Threonine Phosphatases: Mechanism through Structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrak, M.; Kerff, F.; Langsetmo, K.; Tao, T.; Dominguez, R. Structural basis of protein phosphatase 1 regulation. Nature 2004, 429, 780–784. [Google Scholar] [CrossRef] [Green Version]
- Takai, A.; Eto, M.; Hirano, K.; Takeya, K.; Wakimoto, T.; Watanabe, M. Protein phosphatases 1 and 2A and their naturally occurring inhibitors: Current topics in smooth muscle physiology and chemical biology. J. Physiol. Sci 2018, 68, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, D.; Saito-Ito, A.; Asao, N.; Tanabe, K.; Yamamoto, M.; Matsumura, T. Modulation of the growth of Plasmodium falciparum in vitro by protein serine/threonine phosphatase inhibitors. Biochem. Biophys. Res. Commun. 1998, 247, 18–23. [Google Scholar] [CrossRef]
- Chatterjee, J.; Köhn, M. Targeting the untargetable: Recent advances in the selective chemical modulation of protein phosphatase-1 activity. Curr. Opin. Chem. Biol. 2013, 17, 361–368. [Google Scholar] [CrossRef]
- Fontanillo, M.; Köhn, M. Microcystins: Synthesis and structure–activity relationship studies toward PP1 and PP2A. Bioorg Med. Chem. 2018, 26, 1118–1126. [Google Scholar] [CrossRef]
- Woydziak, Z.R.; Yucel, A.J.; Chamberlin, A.R. Tautomycetin Synthetic Analogues: Selective Inhibitors of Protein Phosphatase I. ChemMedChem 2021, 16, 839–850. [Google Scholar] [CrossRef]
- Ammosova, T.; Platonov, M.; Ivanov, A.; Kont, Y.S.; Kumari, N.; Kehn-Hall, K.; Jerebtsova, M.; Kulkarni, A.A.; Uren, A.; Kovalskyy, D.; et al. 1E7-03, a low MW compound targeting host protein phosphatase-1, inhibits HIV-1 transcription. Br. J. Pharm. 2014, 171, 5059–5075. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Ammosova, T.; Choy, M.S.; Pietzsch, C.A.; Ivanov, A.; Ahmad, A.; Saygideğer, Y.; Kumari, N.; Kovalskyy, D.; Üren, A.; et al. Targeting the Non-catalytic RVxF Site of Protein Phosphatase-1 With Small Molecules for Ebola Virus Inhibition. Front. Microbiol. 2019, 10, 2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Kumari, N.; DeMarino, C.; Kont, Y.S.; Ammosova, T.; Kulkarni, A.; Jerebtsova, M.; Vazquez-Meves, G.; Ivanov, A.; Dmytro, K.; et al. Inhibition of HIV-1 infection in humanized mice and metabolic stability of protein phosphatase-1-targeting small molecule 1E7-03. Oncotarget 2017, 8, 76749–76769. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Sajith, A.M.; Wang, S.; Kumari, N.; Choy, M.S.; Ahmad, A.; Cadet, D.R.; Gu, X.; Ivanov, A.I.; Peti, W.; et al. Structural Optimization of 2,3-Dihydro-1H-cyclopenta[b]quinolines Targeting the Noncatalytic RVxF Site of Protein Phosphatase 1 for HIV-1 Inhibition. ACS Infect. Dis. 2020, 6, 3190–3211. [Google Scholar] [CrossRef] [PubMed]
- Trebacz, M.; Wang, Y.; Makotta, L.; Henschke, L.; Köhn, M. Development of a Photoactivatable Protein Phosphatase-1-Disrupting Peptide. J. Org. Chem. 2020, 85, 1712–1717. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Musiyenko, A.; Barik, S. Plasmodium falciparum calcineurin and its association with heat shock protein 90: Mechanisms for the antimalarial activity of cyclosporin A and synergism with geldanamycin. Mol. Biochem. Parasitol. 2005, 141, 29–37. [Google Scholar] [CrossRef]
- Biswas, S.; Saxena, Q.B.; Upender, M. Antimalarial effect of cyclosporin-A on murine P. berghei and human P. falciparum. Indian J. Malariol. 1991, 28, 1–8. [Google Scholar]
- Uadia, P.O.; Ezeamuzie, I.C.; Ladan, M.J.; Gerrets, R. Antimalarial activity of cyclosporins A, C and D. Afr. J. Med. Med. Sci. 1994, 23, 47–51. [Google Scholar]
- Thommen-Scott, K. Antimalarial activity of cyclosporin A. Agents Actions 1981, 11, 770–773. [Google Scholar] [CrossRef]
- Bobbala, D.; Koka, S.; Lang, C.; Boini, K.M.; Huber, S.M.; Lang, F. Effect of cyclosporine on parasitemia and survival of Plasmodium berghei infected mice. Biochem. Biophys. Res. Commun. 2008, 376, 494–498. [Google Scholar] [CrossRef]
- Marin-Menendez, A.; Bell, A. Identification and characterization of novel Plasmodium falciparum cyclophilins and their roles in the antimalarial actions of cyclosporin A and derivatives. Malar. J. 2010, 9, O3. [Google Scholar] [CrossRef] [Green Version]
- Prakash, P.; Zeeshan, M.; Saini, E.; Muneer, A.; Khurana, S.; Kumar Chourasia, B.; Deshmukh, A.; Kaur, I.; Dabral, S.; Singh, N.; et al. Human Cyclophilin B forms part of a multi-protein complex during erythrocyte invasion by Plasmodium falciparum. Nat. Commun. 2017, 8, 1548. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Wali, H.; Jan, S.; Zia, A.; Aslam, M.; Ahmad, I.; Afridi, S.G.; Shams, S.; Khan, A. Analysing the essential proteins set of Plasmodium falciparum PF3D7 for novel drug targets identification against malaria. Malar. J. 2021, 20, 335. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Kumar, R.; Gupta, P.; Mohmmed, A.; Tewari, R.; Malhotra, P.; Gupta, D. High throughput in silico identification and characterization of Plasmodium falciparum PRL phosphatase inhibitors. J. Biomol. Struct. Dyn. 2018, 36, 3531–3540. [Google Scholar] [CrossRef]
- Pandey, R.; Gupta, P.; Mohmmed, A.; Malhotra, P.; Gupta, D. A Plasmodium falciparum protein tyrosine phosphatase inhibitor identified from the ChEMBL-NTD database blocks parasite growth. FEBS Open Bio. 2021, 11, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- PubChem—CID 2928525. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/2928525 (accessed on 1 September 2021).
- RCSB Protein Data Bank—Crystal Structure of PYST1 (MKP3). Available online: https://www.rcsb.org/structure/1MKP (accessed on 1 September 2021).
- Campbell, C.O.; Santiago, D.N.; Guida, W.C.; Manetsch, R.; Adams, J.H. In silico characterization of an atypical MAPK phosphatase of Plasmodium falciparum as a suitable target for drug discovery. Chem. Biol. Drug Des. 2014, 84, 158–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold/Superfamily | Family | Substrate | Number in Each Organism | |||||
|---|---|---|---|---|---|---|---|---|
| Hs | Pf | Pb | Bd | Tg | Cp | |||
| PPPL (Phosphoprotein phosphatases-like) | PPP | pSer/pThr | 13 | 14 | 13 | 6 | 15 | 10 |
| PAP | unknown | 2 | 2 | 2 | 1 | 4 | 2 | |
| PPM (Protein phosphatase Mn2+ or Mg2+-dependent) | PPM | pSer/pThr | 20 | 13 | 13 | 4 | 34 | 10 |
| CC1 (Cysteine-based Class I) | PTP | pTyr | 37 | 2 | 3 | 1 | 1 | 0 |
| DSP | pTyr, pSer/pThr | 40 | 1 | 1 | 1 | 7 | 5 | |
| DSP (RHOD) | pTyr, pSer/pThr | 11 | 2 | 2 | 0 | 4 | 0 | |
| CC2 (Cysteine-based Class II) | LMWPTP | pTyr | 1 | 1 | 1 | 0 | 2 | 2 |
| SSU72 | pSer | 1 | 0 | 0 | 0 | 0 | 0 | |
| CC3 (Cysteine-based Class III) | CDC25 | pTyr, pThr | 3 | 0 | 0 | 0 | 0 | 0 |
| PTPL (Protein tyrosine phosphatase-like) | PTPLA * | 0 | 1 | 1 | 1 | 1 | 1 | |
| HAD (Haloacid dehydrogenase) | EYA | pTyr | 4 | 0 | 0 | 0 | 0 | 0 |
| FCP & NIF-like | pSer | 8 | 4 | 5 | 6 | 9 | 5 | |
| Total | 140 | 40 | 41 | 20 | 77 | 35 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fréville, A.; Gnangnon, B.; Khelifa, A.S.; Gissot, M.; Khalife, J.; Pierrot, C. Deciphering the Role of Protein Phosphatases in Apicomplexa: The Future of Innovative Therapeutics? Microorganisms 2022, 10, 585. https://doi.org/10.3390/microorganisms10030585
Fréville A, Gnangnon B, Khelifa AS, Gissot M, Khalife J, Pierrot C. Deciphering the Role of Protein Phosphatases in Apicomplexa: The Future of Innovative Therapeutics? Microorganisms. 2022; 10(3):585. https://doi.org/10.3390/microorganisms10030585
Chicago/Turabian StyleFréville, Aline, Bénédicte Gnangnon, Asma S. Khelifa, Mathieu Gissot, Jamal Khalife, and Christine Pierrot. 2022. "Deciphering the Role of Protein Phosphatases in Apicomplexa: The Future of Innovative Therapeutics?" Microorganisms 10, no. 3: 585. https://doi.org/10.3390/microorganisms10030585
APA StyleFréville, A., Gnangnon, B., Khelifa, A. S., Gissot, M., Khalife, J., & Pierrot, C. (2022). Deciphering the Role of Protein Phosphatases in Apicomplexa: The Future of Innovative Therapeutics? Microorganisms, 10(3), 585. https://doi.org/10.3390/microorganisms10030585