
Next-Generation High-Throughput Sequencing to Evaluate Bacterial Communities in Freshwater Ecosystem in Hydroelectric Reservoirs


 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
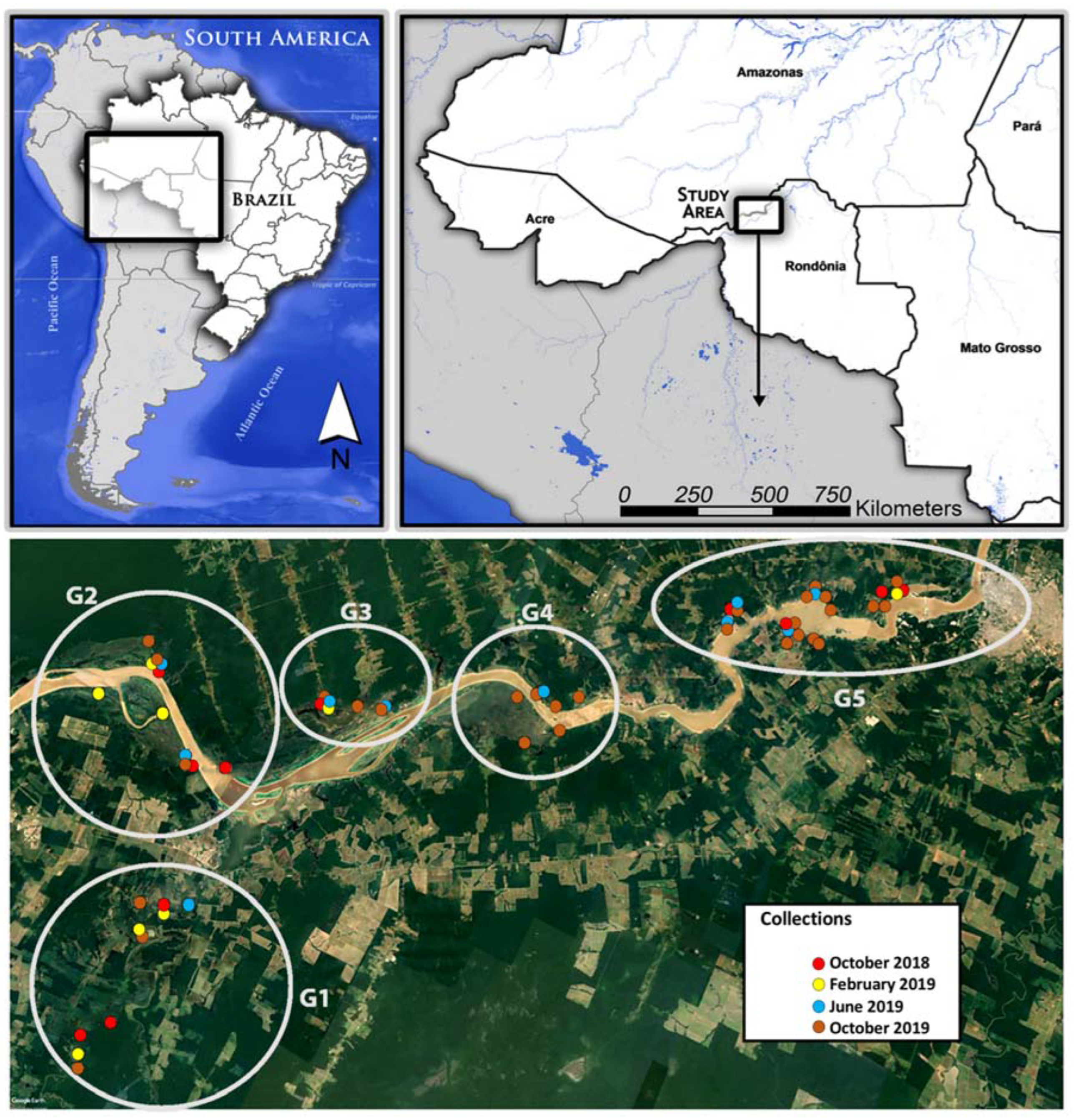
2.1. Study Site
2.2. Sample Collection
2.3. DNA Sample Preparation
2.4. DNA Amplification and Sequencing
2.5. Microbiota Analysis
3. Results
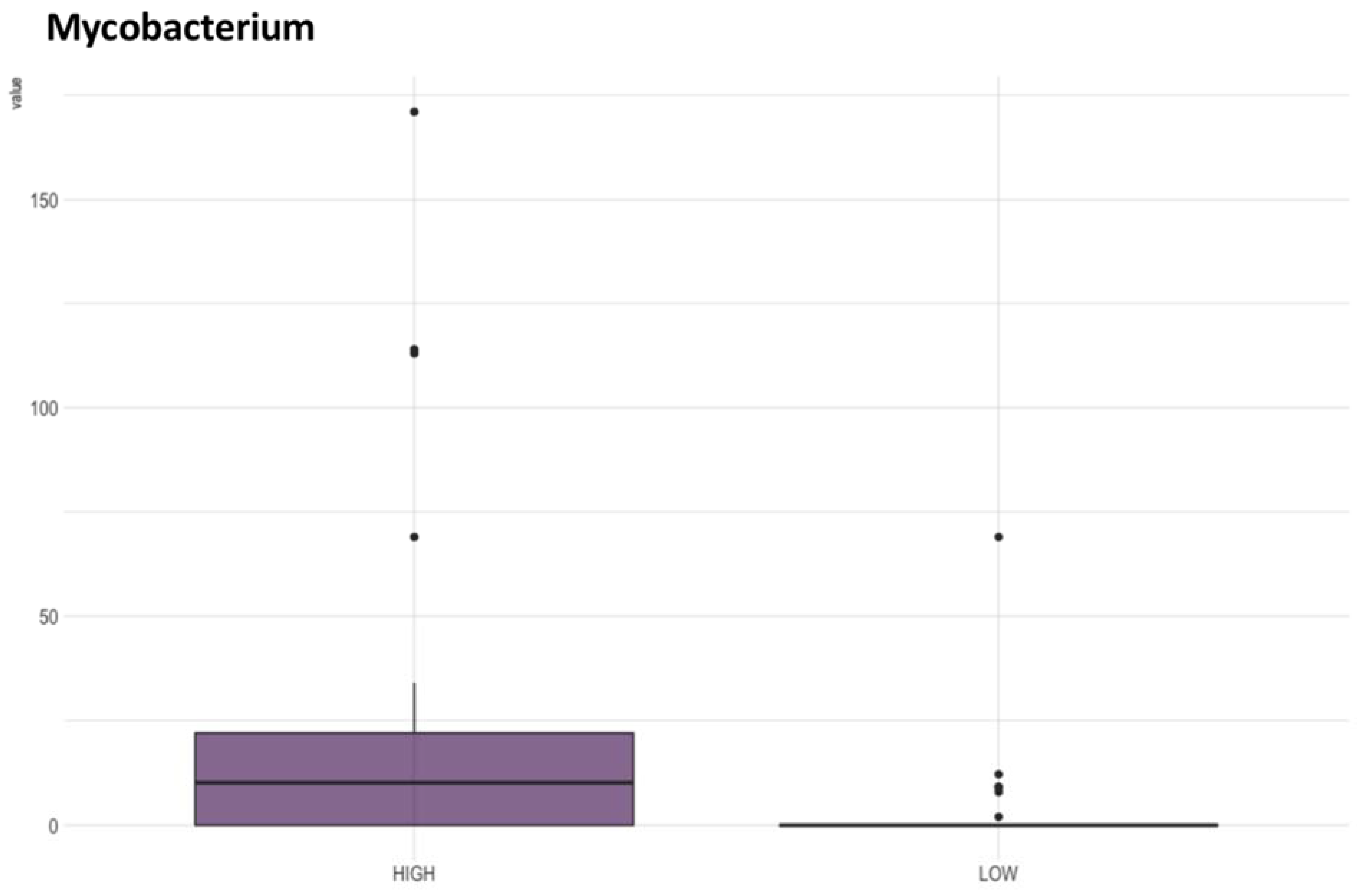
3.1. Analysis of Water Physicochemical Properties versus Microbiome Composition
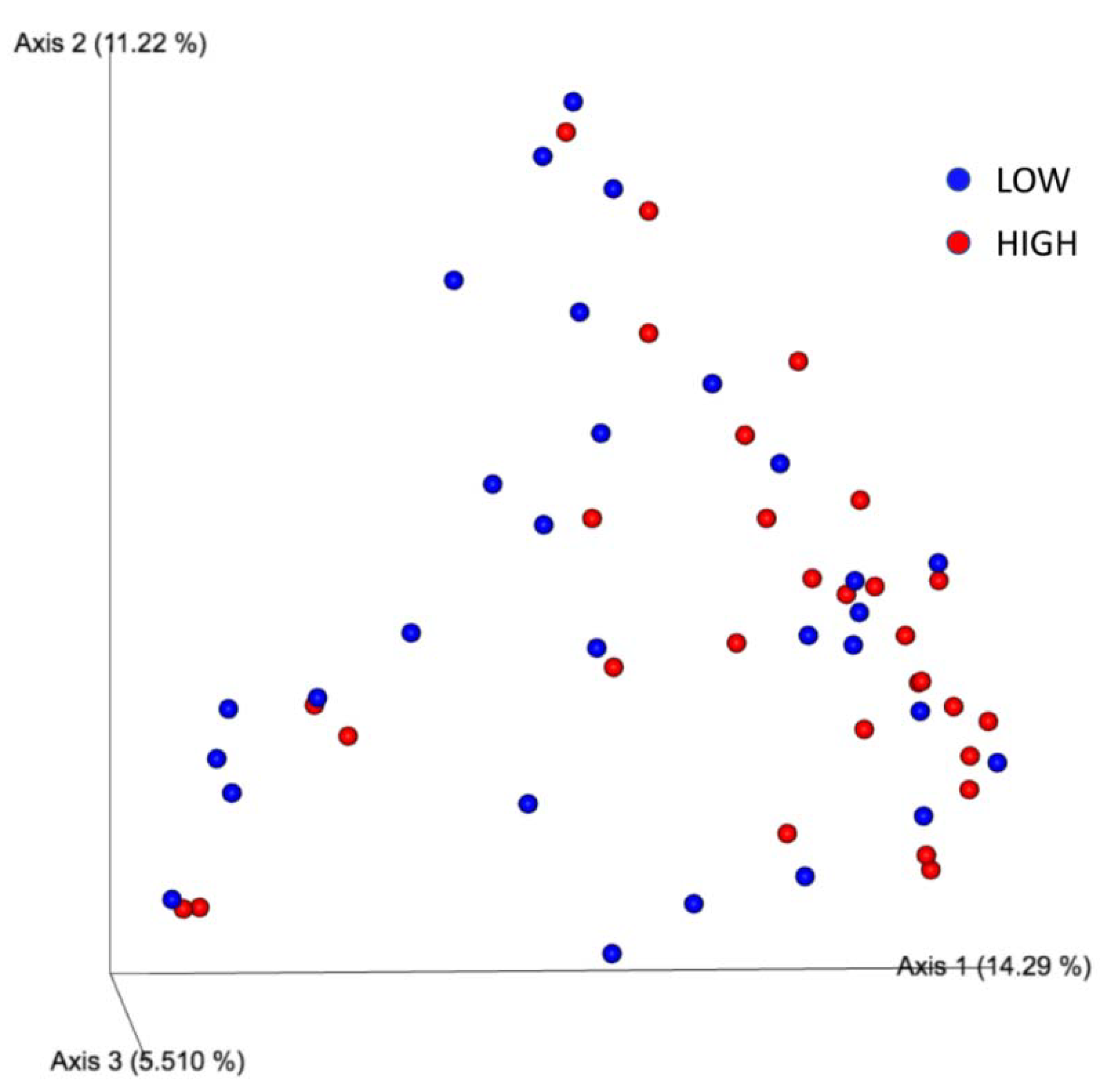
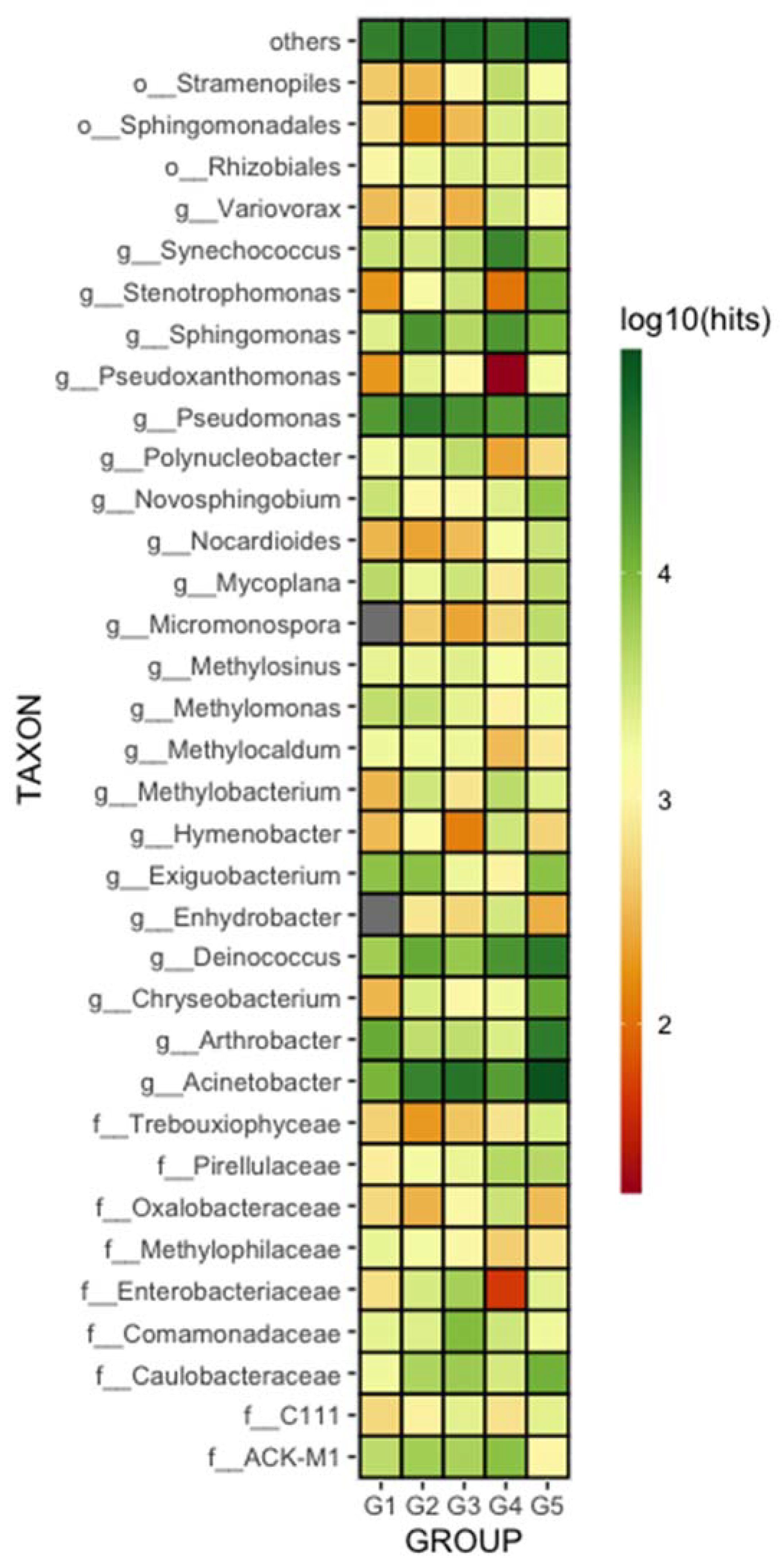
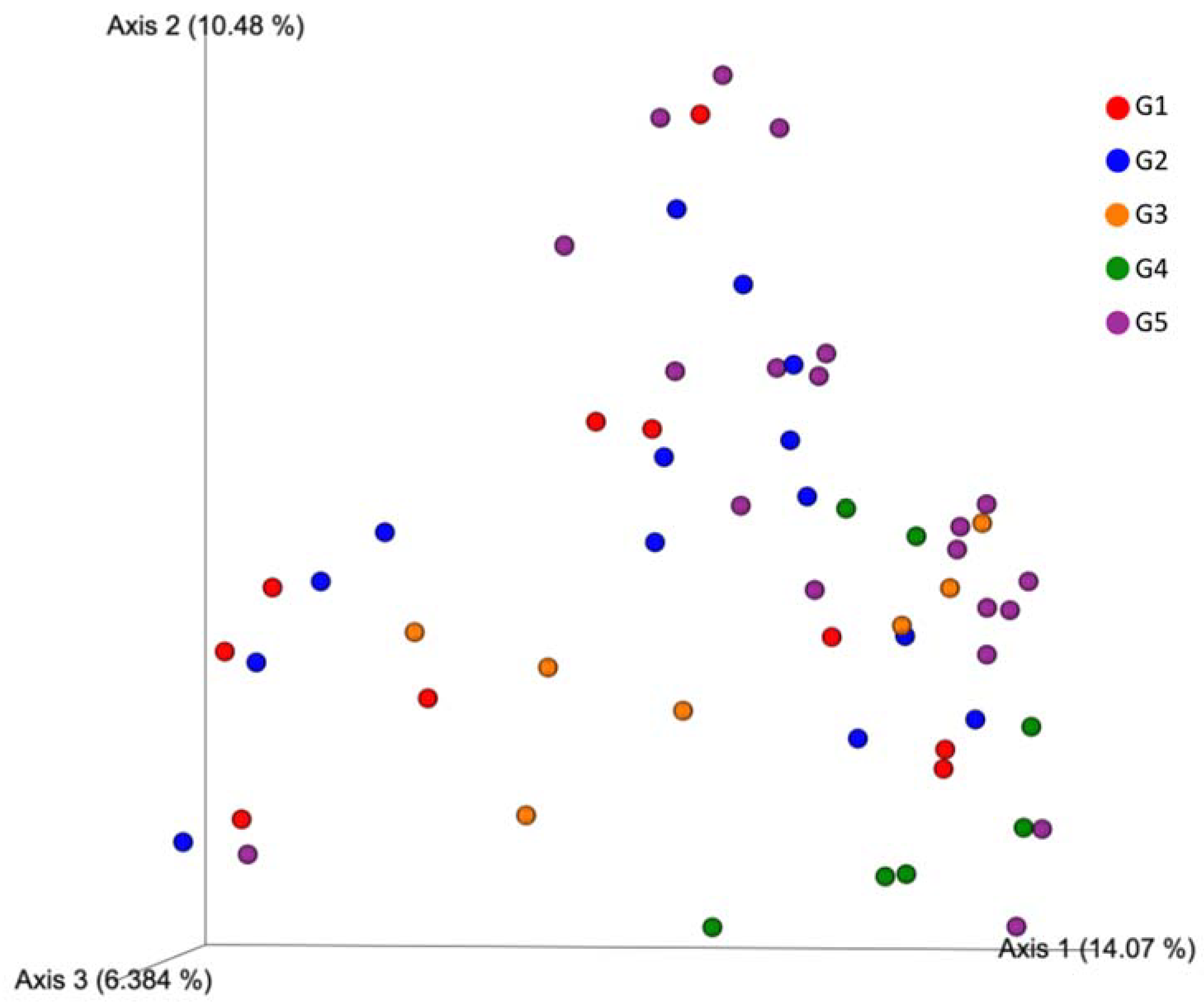
3.2. Geographic Locality versus Microbiome Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Graham, E.B.; Knelman, J.E.; Schindlbacher, A.; Siciliano, S.; Breulmann, M.; Yannarell, A.; Beman, J.M.; Abell, G.; Philippot, L.; Prosser, J.; et al. Microbes as Engines of Ecosystem Function: When Does Community Structure Enhance Predictions of Ecosystem Processes? Front. Microbiol. 2016, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N. Embracing the Unknown: Disentangling the Complexities of the Soil Microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Harwood, V.J.; Staley, C.; Badgley, B.D.; Borges, K.; Korajkic, A. Microbial Source Tracking Markers for Detection of Fecal Contamination in Environmental Waters: Relationships between Pathogens and Human Health Outcomes. FEMS Microbiol. Rev. 2014, 38, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Pachepsky, Y.A.; Allende, A.; Boithias, L.; Cho, K.; Jamieson, R.; Hofstra, N.; Molina, M. Microbial Water Quality: Monitoring and Modeling. J. Environ. Qual. 2018, 47, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Nnadozie, C.F.; Odume, O.N. Freshwater Environments as Reservoirs of Antibiotic Resistant Bacteria and Their Role in the Dissemination of Antibiotic Resistance Genes. Environ. Pollut. 2019, 254, 113067. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, K.; Jun, X.; Bo, L. Role and Functions of Beneficial Microorganisms in Sustainable Aquaculture. Bioresour. Technol. 2009, 100, 3780–3786. [Google Scholar] [CrossRef]
- Pérez-Sánchez, T.; Mora-Sánchez, B.; Balcázar, J.L. Biological Approaches for Disease Control in Aquaculture: Advantages, Limitations and Challenges. Trends Microbiol. 2018, 26, 896–903. [Google Scholar] [CrossRef]
- Bayoumi, H.; Patko, I. Ecological Monitoring of Danube Water Quality in Budapest Region. Am. J. Environ. Sci. 2012, 8, 202–211. [Google Scholar]
- Bonetta, S.; Borelli, E.; Bonetta, S.; Conio, O.; Palumbo, F.; Carraro, E. Development of a PCR Protocol for the Detection of Escherichia coli O157:H7 and Salmonella Spp. in Surface Water. Environ. Monit. Assess. 2011, 177, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Rohr, J.R.; Barrett, C.B.; Civitello, D.J.; Craft, M.E.; Delius, B.; DeLeo, G.A.; Hudson, P.J.; Jouanard, N.; Nguyen, K.H.; Ostfeld, R.S.; et al. Emerging Human Infectious Diseases and the Links to Global Food Production. Nat. Sustain. 2019, 2, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Hammes, F.; Berney, M.; Wang, Y.; Vital, M.; Köster, O.; Egli, T. Flow-Cytometric Total Bacterial Cell Counts as a Descriptive Microbiological Parameter for Drinking Water Treatment Processes. Water Res. 2008, 42, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Dignum, M.; Magic-Knezev, A.; Ross, P.; Rietveld, L.; Hammes, F. Flow Cytometry and Adenosine Tri-Phosphate Analysis: Alternative Possibilities to Evaluate Major Bacteriological Changes in Drinking Water Treatment and Distribution Systems. Water Res. 2012, 46, 4665–4676. [Google Scholar] [CrossRef] [PubMed]
- Zwart, G.; Crump, B.C.; Agterveld, M.P.K.; Hagen, F.; Han, S.K. Typical Freshwater Bacteria: An Analysis of Available 16S rRNA Gene Sequences from Plankton of Lakes and Rivers. Aquat. Microb. Ecol. 2002, 28, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Logares, R.; Bråte, J.; Heinrich, F.; Shalchian-Tabrizi, K.; Bertilsson, S. Infrequent Transitions between Saline and Fresh Waters in One of the Most Abundant Microbial Lineages (SAR11). Mol. Biol. Evol. 2010, 27, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ervinia, A.; Huang, Y. Linking Land Use with Water Pollution in Coastal Watersheds of China. In Challenges towards Ecological Sustainability in China; Springer: Cham, Switzerland, 2019; pp. 241–279. [Google Scholar]
- Yannarell, A.C.; Kent, A.D.; Lauster, G.H.; Kratz, T.K.; Triplett, E.W. Temporal Patterns in Bacterial Communities in Three Temperate Lakes of Different Trophic Status. Microb. Ecol. 2003, 46, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Lindström, E.S.; Kamst-Van Agterveld, M.P.; Zwart, G. Distribution of Typical Freshwater Bacterial Groups Is Associated with pH, Temperature, and Lake Water Retention Time. Appl. Environ. Microbiol. 2005, 71, 8201–8206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yannarell, A.C.; Triplett, E.W. Geographic and Environmental Sources of Variation in Lake Bacterial Community Composition. Appl. Environ. Microbiol. 2005, 71, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Catherine, A.; Troussellier, M.; Bernard, C. Design and Application of a Stratified Sampling Strategy to Study the Regional Distribution of Cyanobacteria (Ile-de-France, France). Water Res. 2008, 42, 4989–5001. [Google Scholar] [CrossRef] [Green Version]
- Marmen, S.; Blank, L.; Al-Ashhab, A.; Malik, A.; Ganzert, L.; Lalzar, M.; Grossart, H.-P.; Sher, D. The Role of Land Use Types and Water Chemical Properties in Structuring the Microbiomes of a Connected Lake System. Front. Microbiol. 2020, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Chavarria, K.A.; Saltonstall, K.; Vinda, J.; Batista, J.; Lindmark, M.; Stallard, R.F.; Hall, J.S. Land Use Influences Stream Bacterial Communities in Lowland Tropical Watersheds. Sci. Rep. 2021, 11, 21752. [Google Scholar] [CrossRef]
- Chavarria, R.A.; Game, M.; Arbelaez, B.; Ramnarine, C.; Snow, Z.K.; Smith, F.W. Extensive Loss of Wnt Genes in Tardigrada. BMC Ecol. Evol. 2021, 21, 223. [Google Scholar] [CrossRef] [PubMed]
- Garner, E.; Davis, B.C.; Milligan, E.; Blair, M.F.; Keenum, I.; Maile-Moskowitz, A.; Pan, J.; Gnegy, M.; Liguori, K.; Gupta, S.; et al. Next Generation Sequencing Approaches to Evaluate Water and Wastewater Quality. Water Res. 2021, 194, 116907. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R.; Rodriguez-Valera, F.; McMahon, K.D.; Toyama, D.; Rinke, R.; Cristina Souza de Oliveira, T.; Wagner Garcia, J.; Pellon de Miranda, F.; Henrique-Silva, F. Metagenomics of the Water Column in the Pristine Upper Course of the Amazon River. PLoS ONE 2011, 6, e23785. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, K.D.; Villegas, L.E.M.; Fernandes, G.R.; David, M.R.; Maciel-de-Freitas, R.; Moreira, L.A.; Lorenzo, M.G. Egg-laying by female Aedes aegypti shapes the bacterial communities of breeding sites. bioRxiv 2022. [Google Scholar] [CrossRef]
- Wong, J.; Stoddard, S.T.; Astete, H.; Morrison, A.C.; Scott, T.W. Oviposition Site Selection by the Dengue Vector Aedes Aegypti and Its Implications for Dengue Control. PLoS Negl. Trop. Dis. 2011, 5, e1015. [Google Scholar] [CrossRef]
- Onchuru, T.O.; Ajamma, Y.U.; Burugu, M.; Kaltenpoth, M.; Masiga, D.; Villinger, J. Chemical Parameters and Bacterial Communities Associated with Larval Habitats of Anopheles, Culex and Aedes Mosquitoes (Diptera: Culicidae) in Western Kenya. Int. J. Trop. Insect Sci. 2016, 36, 146–160. [Google Scholar] [CrossRef]
- Kroth, N.; Cozzer, G.D.; de Carvalho, G.; Cassol, A.S.; Breaux, J.; Lutinski, J.A.; Busato, M.A.; Roman Junior, W.A.; Dos Santos, J.J.; Albeny-Simões, D. Oviposition Preferences of the Mosquito Linnaeus, 1762 (Culicidae): An Urban Environment Bioassay. Bull. Entomol. Res. 2019, 109, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Hery, L.; Guidez, A.; Durand, A.-A.; Delannay, C.; Normandeau-Guimond, J.; Reynaud, Y.; Issaly, J.; Goindin, D.; Legrave, G.; Gustave, J.; et al. Natural Variation in Physicochemical Profiles and Bacterial Communities Associated with Aedes Aegypti Breeding Sites and Larvae on Guadeloupe and French Guiana. Microb. Ecol. 2021, 81, 93–109. [Google Scholar] [CrossRef]
- Ponnusamy, L.; Xu, N.; Nojima, S.; Wesson, D.M.; Schal, C.; Apperson, C.S. Identification of Bacteria and Bacteria-Associated Chemical Cues That Mediate Oviposition Site Preferences by Aedes Aegypti. Proc. Natl. Acad. Sci. USA 2008, 105, 9262–9267. [Google Scholar] [CrossRef] [Green Version]
- Melo, N.; Wolff, G.H.; Costa-da-Silva, A.L.; Arribas, R.; Triana, M.F.; Gugger, M.; Riffell, J.A.; DeGennaro, M.; Stensmyr, M.C. Geosmin Attracts Aedes Aegypti Mosquitoes to Oviposition Sites. Curr. Biol. 2020, 30, 127–134.e5. [Google Scholar] [CrossRef] [Green Version]
- Alto, B.W.; Philip Lounibos, L. Vector Competence for Arboviruses in Relation to the Larval Environment of Mosquitoes. In Ecology of Parasite-Vector Interactions; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 81–101. [Google Scholar]
- Jupatanakul, N.; Sim, S.; Dimopoulos, G. The Insect Microbiome Modulates Vector Competence for Arboviruses. Viruses 2014, 6, 4294–4313. [Google Scholar] [CrossRef] [PubMed]
- Souza-Neto, J.A.; Powell, J.R.; Bonizzoni, M. Aedes Aegypti Vector Competence Studies: A Review. Infect. Genet. Evol. 2019, 67, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Ab’saber, A.N. Domínios de Natureza no Brasil, Os—Potencialidades Paisagísticas; Atelie Editorial: Cotia, Brazil, 2012; ISBN 9788574805962. [Google Scholar]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- de Berardinis, V.; Durot, M.; Weissenbach, J.; Salanoubat, M. Acinetobacter Baylyi ADP1 as a Model for Metabolic System Biology. Curr. Opin. Microbiol. 2009, 12, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Mahjoubi, M.; Jaouani, A.; Guesmi, A.; Ben Amor, S.; Jouini, A.; Cherif, H.; Najjari, A.; Boudabous, A.; Koubaa, N.; Cherif, A. Hydrocarbonoclastic Bacteria Isolated from Petroleum Contaminated Sites in Tunisia: Isolation, Identification and Characterization of the Biotechnological Potential. New Biotechnol. 2013, 30, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Tazaki, K.; Minamikawa, T. Occurrence of Shikimic and Quinic Acids in Angiosperms. Phytochemistry 1975, 14, 195–197. [Google Scholar] [CrossRef]
- Daly, M.J. Modulating Radiation Resistance: Insights Based on Defenses against Reactive Oxygen Species in the Radioresistant Bacterium Deinococcus radiodurans. Clin. Lab. Med. 2006, 26, 491–504. [Google Scholar] [CrossRef]
- Jin, M.; Xiao, A.; Zhu, L.; Zhang, Z.; Huang, H.; Jiang, L. The Diversity and Commonalities of the Radiation-Resistance Mechanisms of Deinococcus and Its up-to-Date Applications. AMB Express 2019, 9, 138. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Baldini, R.L.; Déziel, E.; Saucier, M.; Zhang, Q.; Liberati, N.T.; Lee, D.; Urbach, J.; Goodman, H.M.; Rahme, L.G. The Broad Host Range Pathogen Pseudomonas aeruginosa Strain PA14 Carries Two Pathogenicity Islands Harboring Plant and Animal Virulence Genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2530–2535. [Google Scholar] [CrossRef] [Green Version]
- Özen, A.I.; Ussery, D.W. Defining the Pseudomonas Genus: Where Do We Draw the Line with Azotobacter? Microb. Ecol. 2012, 63, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Newton, R.J.; Jones, S.E.; Eiler, A.; McMahon, K.D.; Bertilsson, S. A Guide to the Natural History of Freshwater Lake Bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 14–49. [Google Scholar] [CrossRef] [Green Version]
- Morris, C.E.; Sands, D.C.; Vinatzer, B.A.; Glaux, C.; Guilbaud, C.; Buffière, A.; Yan, S.; Dominguez, H.; Thompson, B.M. The Life History of the Plant Pathogen Pseudomonas syringae Is Linked to the Water Cycle. ISME J. 2008, 2, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, R.B.; Vinatzer, B.A.; Schmale, D.G. Diversity and Abundance of Ice Nucleating Strains of Pseudomonas syringae in a Freshwater Lake in Virginia, USA. Front. Microbiol. 2017, 8, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, C.E.; Conen, F.; Alex Huffman, J.; Phillips, V.; Pöschl, U.; Sands, D.C. Bioprecipitation: A Feedback Cycle Linking Earth History, Ecosystem Dynamics and Land Use through Biological Ice Nucleators in the Atmosphere. Glob. Chang. Biol. 2014, 20, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.J.; Sharpley, A.N.; Ahuja, L.R. Agricultural Chemical Discharge in Surface Water Runoff. J. Environ. Qual. 1993, 22, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Montuelle, B.; Dorigo, U.; Bérard, A.; Volat, B.; Bouchez, A.; Tlili, A.; Gouy, V.; Pesce, S. The Periphyton as a Multimetric Bioindicator for Assessing the Impact of Land Use on Rivers: An Overview of the Ardières-Morcille Experimental Watershed (France). Hydrobiologia 2010, 657, 123–141. [Google Scholar] [CrossRef]
- Zeglin, L.H. Stream Microbial Diversity in Response to Environmental Changes: Review and Synthesis of Existing Research. Front. Microbiol. 2015, 6, 454. [Google Scholar] [CrossRef] [Green Version]
- Saxton, M.A.; Morrow, E.A.; Bourbonniere, R.A.; Wilhelm, S.W. Glyphosate Influence on Phytoplankton Community Structure in Lake Erie. J. Great Lakes Res. 2011, 37, 683–690. [Google Scholar] [CrossRef]
- Sorokin, Y.I.; Zakuskina, O.Y. Features of the Comacchio Ecosystem Transformed during Persistent Bloom of Picocyanobacteria. J. Oceanogr. 2010, 66, 373–387. [Google Scholar] [CrossRef]
- Li, J.; Chen, Z.; Jing, Z.; Zhou, L.; Li, G.; Ke, Z.; Jiang, X.; Liu, J.; Liu, H.; Tan, Y. Synechococcus Bloom in the Pearl River Estuary and Adjacent Coastal Area-With Special Focus on Flooding during Wet Seasons. Sci. Total Environ. 2019, 692, 769–783. [Google Scholar] [CrossRef]
- Bubak, I.; Śliwińska-Wilczewska, S.; Głowacka, P.; Szczerba, A.; Możdżeń, K. The Importance of Allelopathic Picocyanobacterium Sp. on the Abundance, Biomass Formation, and Structure of Phytoplankton Assemblages in Three Freshwater Lakes. Toxins 2020, 12, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedes, I.A.; Rachid, C.T.C.; Rangel, L.M.; Silva, L.H.S.; Bisch, P.M.; Azevedo, S.M.F.; Pacheco, A.B.F. Close Link Between Harmful Cyanobacterial Dominance and Associated Bacterioplankton in a Tropical Eutrophic Reservoir. Front. Microbiol. 2018, 9, 424. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.; Fujiwara, N.; Naka, T.; Mitani, A.; Kubota, H.; Tomida, J.; Morita, Y.; Hitomi, J. Genus Enhydrobacter Staley et Al. 1987 Should Be Recognized as a Member of the Family Rhodospirillaceae within the Class Alphaproteobacteria. Microbiol. Immunol. 2012, 56, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Staley, J.T.; Brenner, D.J. Enhydrobacter. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–3. [Google Scholar]
- Wang, H.; Zhang, W.; Ye, Y.; He, Q.; Zhang, S. Isolation and Characterization of Pseudoxanthomonas sp. Strain YP1 Capable of Denitrifying Phosphorus Removal (DPR). Geomicrobiol. J. 2018, 35, 537–543. [Google Scholar] [CrossRef]
- Rajta, A.; Bhatia, R.; Setia, H.; Pathania, P. Role of Heterotrophic Aerobic Denitrifying Bacteria in Nitrate Removal from Wastewater. J. Appl. Microbiol. 2020, 128, 1261–1278. [Google Scholar] [CrossRef]
- Hu, X.; Yu, J.; Wang, C.; Chen, H. Cellulolytic Bacteria Associated with the Gut of Dendroctonus Armandi Larvae (Coleoptera: Curculionidae: Scolytinae). Forests 2014, 5, 455–465. [Google Scholar] [CrossRef]
- Borges, A.V.; Darchambeau, F.; Lambert, T.; Bouillon, S.; Morana, C.; Brouyère, S.; Hakoun, V.; Jurado, A.; Tseng, H.-C.; Descy, J.-P.; et al. Effects of Agricultural Land Use on Fluvial Carbon Dioxide, Methane and Nitrous Oxide Concentrations in a Large European River, the Meuse (Belgium). Sci. Total Environ. 2018, 610–611, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Ozbayram, E.G.; Gozde Ozbayram, E.; Koker, L.; Akçaalan, R.; Aydın, F.; Ertürk, A.; Ince, O.; Albay, M. Contrasting the Water Quality and Bacterial Community Patterns in Shallow and Deep Lakes: Manyas vs. Iznik. Environ. Manag. 2021, 67, 506–512. [Google Scholar] [CrossRef]
- Le, H.T.; Ho, C.T.; Trinh, Q.H.; Trinh, D.A.; Luu, M.T.N.; Tran, H.S.; Orange, D.; Janeau, J.L.; Merroune, A.; Rochelle-Newall, E.; et al. Responses of Aquatic Bacteria to Terrestrial Runoff: Effects on Community Structure and Key Taxonomic Groups. Front. Microbiol. 2016, 7, 889. [Google Scholar] [CrossRef] [Green Version]
- Staley, Z.R.; Chase, E.; Mitraki, C.; Crisman, T.L.; Harwood, V.J. Microbial Water Quality in Freshwater Lakes with Different Land Use. J. Appl. Microbiol. 2013, 115, 1240–1250. [Google Scholar] [CrossRef]
- Staley, C.; Gould, T.J.; Wang, P.; Phillips, J.; Cotner, J.B.; Sadowsky, M.J. Core Functional Traits of Bacterial Communities in the Upper Mississippi River Show Limited Variation in Response to Land Cover. Front. Microbiol. 2014, 5, 414. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Johnson, D.; Gould, T.J.; Wang, P.; Phillips, J.; Cotner, J.B.; Sadowsky, M.J. Frequencies of Heavy Metal Resistance Are Associated with Land Cover Type in the Upper Mississippi River. Sci. Total Environ. 2015, 511, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Niño-García, J.P.; Ruiz-González, C.; Del Giorgio, P.A. Interactions between Hydrology and Water Chemistry Shape Bacterioplankton Biogeography across Boreal Freshwater Networks. ISME J. 2016, 10, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, D.T.; Jovanovic, D.; Lintern, A.; Teakle, I.; Barnes, M.; Deletic, A.; Coleman, R.; Rooney, G.; Prosser, T.; Coutts, S.; et al. Source Tracking Using Microbial Community Fingerprints: Method Comparison with Hydrodynamic Modelling. Water Res. 2017, 109, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-Y.; Sudduth, E.B.; Wallenstein, M.D.; Wright, J.P.; Bernhardt, E.S. Watershed Urbanization Alters the Composition and Function of Stream Bacterial Communities. PLoS ONE 2011, 6, e22972. [Google Scholar] [CrossRef]
- Garrido, L.; Sánchez, O.; Ferrera, I.; Tomàs, N.; Mas, J. Dynamics of Microbial Diversity Profiles in Waters of Different Qualities. Approximation to an Ecological Quality Indicator. Sci. Total Environ. 2014, 468–469, 1154–1161. [Google Scholar] [CrossRef]
- Van Rossum, T.; Peabody, M.A.; Uyaguari-Diaz, M.I.; Cronin, K.I.; Chan, M.; Slobodan, J.R.; Nesbitt, M.J.; Suttle, C.A.; Hsiao, W.W.L.; Tang, P.K.C.; et al. Year-Long Metagenomic Study of River Microbiomes Across Land Use and Water Quality. Front. Microbiol. 2015, 6, 1405. [Google Scholar] [CrossRef] [Green Version]
- Delghandi, M.R.; Waldner, K.; El-Matbouli, M.; Menanteau-Ledouble, S. Identification Mycobacterium Spp. in the Natural Water of Two Austrian Rivers. Microorganisms 2020, 8, 1305. [Google Scholar] [CrossRef]
- Bartram, J.; Cotruvo, J.; Dufour, A.; Rees, G.; Pedley, S. Pathogenic Mycobacteria in Water: A Guide to Public Health Consequences, Monitoring and Management; World Health Organization: Geneva, Switzerland, 2004; ISBN 9789241562591. [Google Scholar]
- Blanc, S.M.; Robinson, D.; Fahrenfeld, N.L. Potential for Nontuberculous Mycobacteria Proliferation in Natural and Engineered Water Systems due to Climate Change: A Literature Review. City Environ. Interact. 2021, 11, 100070. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | ||||||||
| Sample Size | Permutations | Pseudo-F | p-Value | q-Value | ||||
| Group 1 | Group 2 | |||||||
| G1 | G2 | 21 | 999 | 0.801 | 0.720 | 0.720 | ||
| G3 | 17 | 999 | 1.243 | 0.166 | 0.184 | |||
| G4 | 17 | 999 | 2.386 | 0.002 | 0.010 | |||
| G5 | 33 | 999 | 1.686 | 0.038 | 0.054 | |||
| G2 | G3 | 18 | 999 | 1.532 | 0.033 | 0.054 | ||
| G4 | 18 | 999 | 2.992 | 0.001 | 0.010 | |||
| G5 | 34 | 999 | 1.892 | 0.012 | 0.024 | |||
| G3 | G4 | 14 | 999 | 1.671 | 0.010 | 0.024 | ||
| G5 | 30 | 999 | 1.409 | 0.068 | 0.085 | |||
| G4 | G5 | 30 | 999 | 2.084 | 0.007 | 0.023 | ||
| (b) | ||||||||
| Sample size | Permutations | pseudo-F | p-value | q-value | ||||
| Group 1 | Group 2 | |||||||
| HIGH | LOW | 58 | 999 | 1.440455 | 0.057 | 0.057 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, M.V.R.; Alonso, D.P.; Dropa, M.; Razzolini, M.T.P.; de Carvalho, D.P.; Ribeiro, K.A.N.; Ribolla, P.E.M.; Sallum, M.A.M. Next-Generation High-Throughput Sequencing to Evaluate Bacterial Communities in Freshwater Ecosystem in Hydroelectric Reservoirs. Microorganisms 2022, 10, 1398. https://doi.org/10.3390/microorganisms10071398
Rojas MVR, Alonso DP, Dropa M, Razzolini MTP, de Carvalho DP, Ribeiro KAN, Ribolla PEM, Sallum MAM. Next-Generation High-Throughput Sequencing to Evaluate Bacterial Communities in Freshwater Ecosystem in Hydroelectric Reservoirs. Microorganisms. 2022; 10(7):1398. https://doi.org/10.3390/microorganisms10071398
Chicago/Turabian StyleRojas, Martha Virginia R., Diego Peres Alonso, Milena Dropa, Maria Tereza P. Razzolini, Dario Pires de Carvalho, Kaio Augusto Nabas Ribeiro, Paulo Eduardo M. Ribolla, and Maria Anice M. Sallum. 2022. "Next-Generation High-Throughput Sequencing to Evaluate Bacterial Communities in Freshwater Ecosystem in Hydroelectric Reservoirs" Microorganisms 10, no. 7: 1398. https://doi.org/10.3390/microorganisms10071398
APA StyleRojas, M. V. R., Alonso, D. P., Dropa, M., Razzolini, M. T. P., de Carvalho, D. P., Ribeiro, K. A. N., Ribolla, P. E. M., & Sallum, M. A. M. (2022). Next-Generation High-Throughput Sequencing to Evaluate Bacterial Communities in Freshwater Ecosystem in Hydroelectric Reservoirs. Microorganisms, 10(7), 1398. https://doi.org/10.3390/microorganisms10071398