

Hepatitis B Virus Genotype D: An Overview of Molecular Epidemiology, Evolutionary History, and Clinical Characteristics


Abstract
:1. Introduction
2. Genotype D
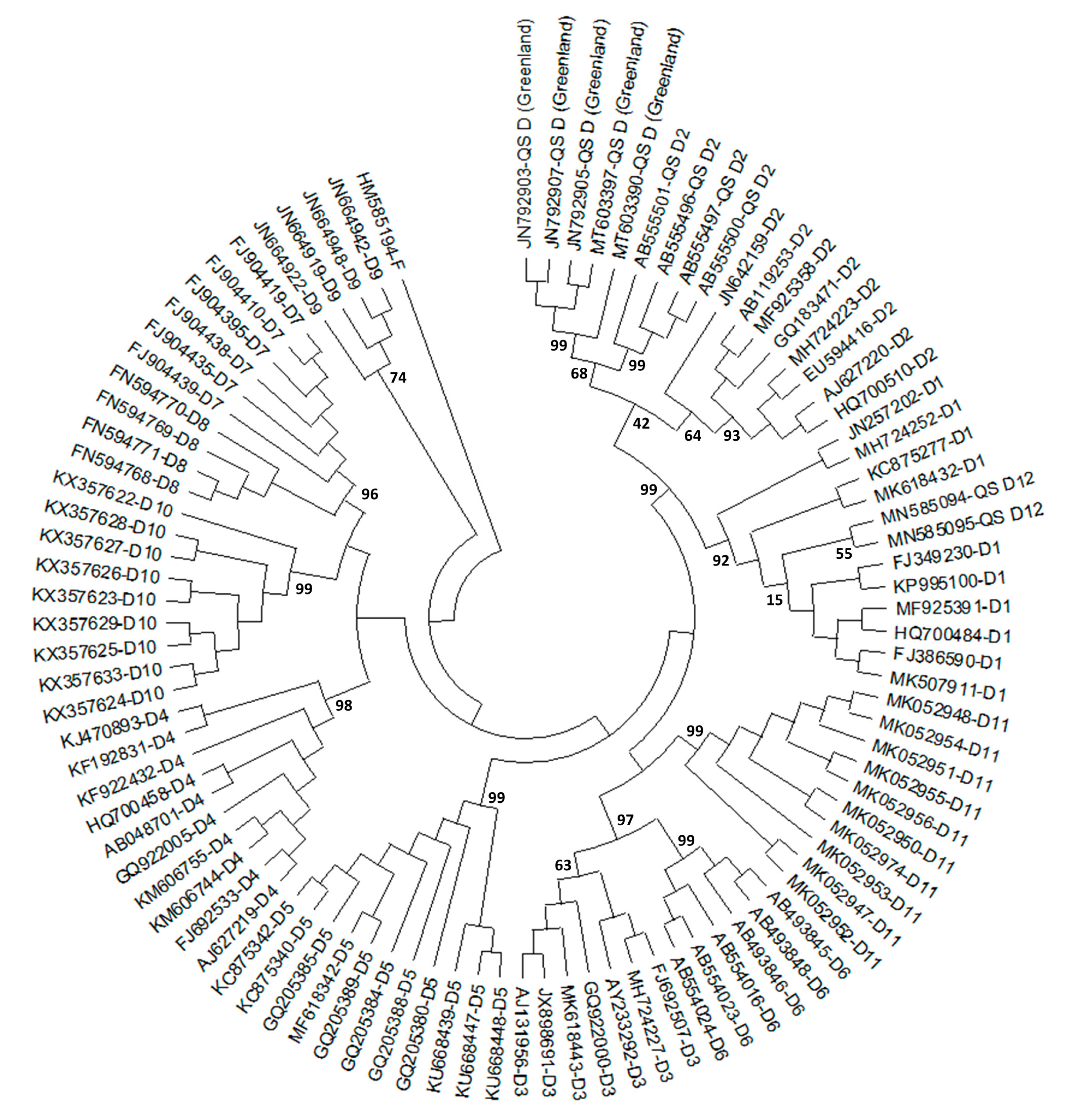
2.1. History of HBV/D Subgenotyping
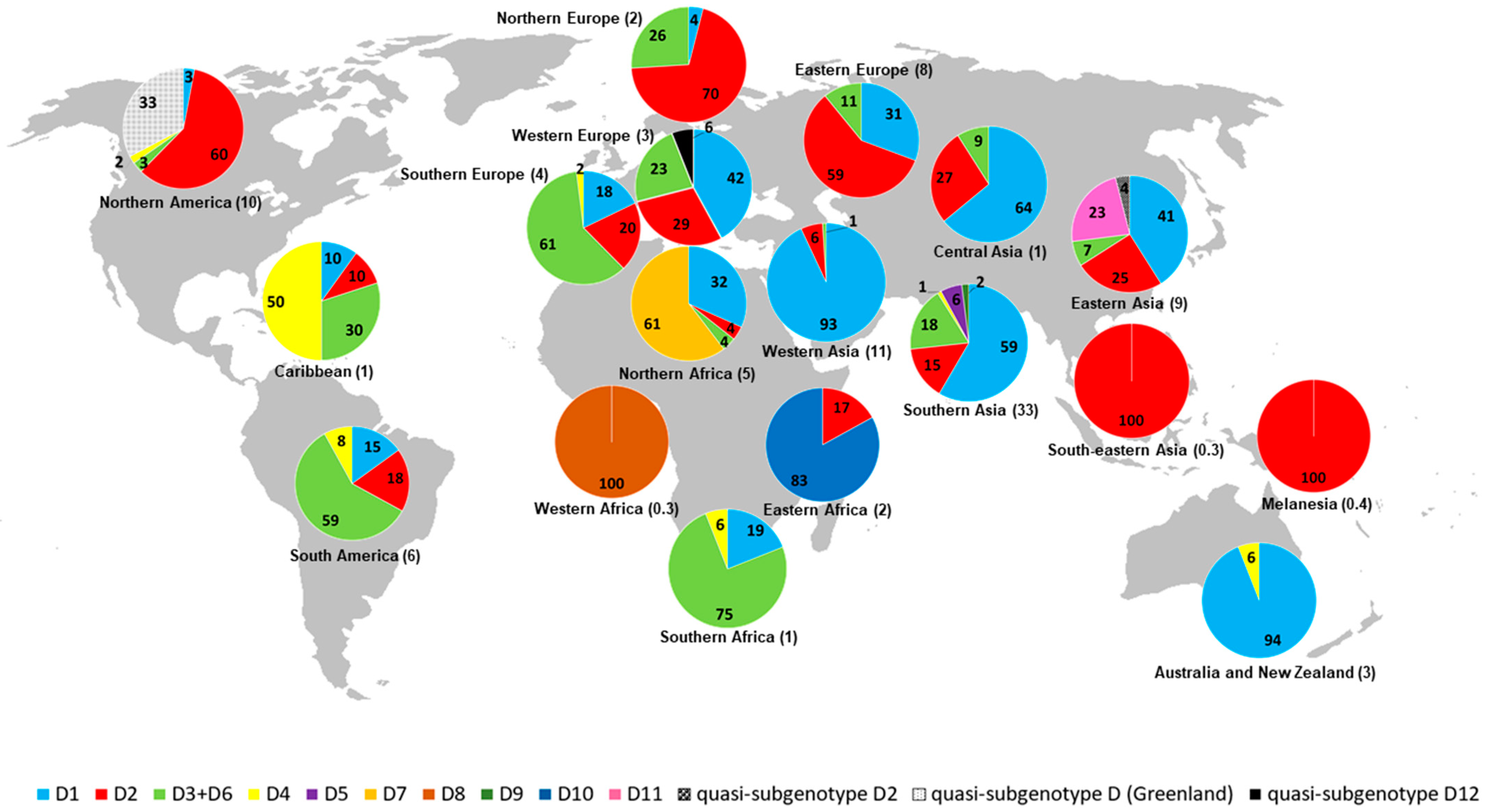
2.2. Geographic Distribution of HBV/D Subgenotypes
2.3. Origin and Worldwide Spread of HBV/D
2.4. Clinical Associations of HBV/D Subgenotypes with Infection
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. World Health Organization. Fact Sheets. Hepatitis B. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 3 January 2023).
- Trépo, C.; Chan, H.L.Y.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.-M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on Prevention, Diagnosis, and Treatment of Chronic Hepatitis B: AASLD 2018 Hepatitis B Guidance. Clin. Liver Dis. 2018, 12, 33–34. [Google Scholar] [CrossRef] [Green Version]
- Araujo, N.M.; Waizbort, R.; Kay, A. Hepatitis B virus infection from an evolutionary point of view: How viral, host, and environmental factors shape genotypes and subgenotypes. Infect. Genet. Evol. 2011, 11, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Zoulim, F.; Mason, W.S. Hepadnaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2185–2221. [Google Scholar]
- Kay, A.; Zoulim, F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007, 127, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Araujo, N.M. Hepatitis B virus intergenotypic recombinants worldwide: An overview. Infect. Genet. Evol. 2015, 36, 500–510. [Google Scholar] [CrossRef]
- Simmonds, P.; Midgley, S. Recombination in the Genesis and Evolution of Hepatitis B Virus Genotypes. J. Virol. 2005, 79, 15467–15476. [Google Scholar] [CrossRef] [Green Version]
- Kramvis, A. Genotypes and Genetic Variability of Hepatitis B Virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef]
- Kramvis, A.; Arakawa, K.; Yu, M.C.; Nogueira, R.; Stram, D.O.; Kew, M.C. Relationship of serological subtype, basic core promoter and precore mutations to genotypes/subgenotypes of hepatitis B virus. J. Med. Virol. 2008, 80, 27–46. [Google Scholar] [CrossRef]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Kurbanov, F.; Van Ranst, M.; Tacke, F. Molecular identification of hepatitis B virus genotypes/subgenotypes: Revised classification hurdles and updated resolutions. World J. Gastroenterol. 2014, 20, 7152–7168. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhang, Z.; Ling, C.; Zheng, W.; Zhu, C.-D.; Carr, M.; Higgins, D. Hepatitis B virus subgenotyping: History, effects of recombination, misclassifications, and corrections. Infect. Genet. Evol. 2013, 16, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Araujo, N.M.; Teles, S.A.; Spitz, N. Comprehensive Analysis of Clinically Significant Hepatitis B Virus Mutations in Relation to Genotype, Subgenotype and Geographic Region. Front. Microbiol. 2020, 11, 616023. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-J.; Lok, A.S.F. Clinical significance of hepatitis B virus genotypes. Hepatology 2002, 35, 1274–1276. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.K.; Revill, P.A.; Ahn, S.H. HBV Genotypes: Relevance to natural history, pathogenesis and treatment of Chronic Hepatitis B. Antivir. Ther. 2011, 16, 1169–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-L.; Kao, J.-H. The clinical implications of hepatitis B virus genotype: Recent advances. J. Gastroenterol. Hepatol. 2011, 26, 123–130. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.J. The influence of hepatitis B virus genotype and subgenotype on the natural history of chronic hepatitis B. Hepatol. Int. 2009, 3, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocher, A.; Papac, L.; Barquera, R.; Key, F.M.; Spyrou, M.A.; Hübler, R.; Rohrlach, A.B.; Aron, F.; Stahl, R.; Wissgott, A.; et al. Ten millennia of hepatitis B virus evolution. Science 2021, 374, 182–188. [Google Scholar] [CrossRef]
- Kostaki, E.-G.; Karamitros, T.; Stefanou, G.; Mamais, I.; Angelis, K.; Hatzakis, A.; Kramvis, A.; Paraskevis, D. Unravelling the history of hepatitis B virus genotypes A and D infection using a full-genome phylogenetic and phylogeographic approach. Elife 2018, 7, e36709. [Google Scholar] [CrossRef]
- Krause-Kyora, B.; Susat, J.; Key, F.M.; Kühnert, D.; Bosse, E.; Immel, A.; Rinne, C.; Kornell, S.-C.; Yepes, D.; Franzenburg, S.; et al. Neolithic and medieval virus genomes reveal complex evolution of hepatitis B. Elife 2018, 7, e36666. [Google Scholar] [CrossRef]
- Mühlemann, B.; Jones, T.C.; Damgaard, P.D.B.; Allentoft, M.E.; Shevnina, I.; Logvin, A.; Usmanova, E.; Panyushkina, I.P.; Boldgiv, B.; Bazartseren, T.; et al. Ancient hepatitis B viruses from the Bronze Age to the Medieval period. Nature 2018, 557, 418–423. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, H.; Tsuda, F.; Sakugawa, H.; Sastrosoewignjo, R.I.; Imai, M.; Miyakawa, Y.; Mayumi, M. Typing Hepatitis B Virus by homology in nucleotide sequence: Comparison of surface antigen subtypes. J. Gen. Virol. 1988, 69, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Yousif, M.; Kramvis, A. Genotype D of hepatitis B virus and its subgenotypes: An update. Hepatol. Res. 2013, 43, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Norder, H.; Couroucé, A.-M.; Coursaget, P.; Echevarria, J.M.; Lee, S.-D.; Mushahwar, I.K.; Robertson, B.H.; Locarnini, S.; Magnius, L.O. Genetic Diversity of Hepatitis B Virus Strains Derived Worldwide: Genotypes, Subgenotypes, and HBsAg Subtypes. Intervirology 2004, 47, 289–309. [Google Scholar] [CrossRef]
- Banerjee, A.; Kurbanov, F.; Datta, S.; Chandra, P.K.; Tanaka, Y.; Mizokami, M.; Chakravarty, R. Phylogenetic relatedness and genetic diversity of hepatitis B virus isolates in Eastern India. J. Med. Virol. 2006, 78, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Lusida, M.I.; Nugrahaputra, V.E.; Soetjipto Handajani, R.; Nagano-Fujii, M.; Sasayama, M.; Utsumi, T.; Hotta, H. Novel Subgenotypes of Hepatitis B Virus Genotypes C and D in Papua, Indonesia. J. Clin. Microbiol. 2008, 46, 2160–2166. [Google Scholar] [CrossRef] [Green Version]
- Utsumi, T.; Lusida, M.I.; Yano, Y.; Nugrahaputra, V.E.; Amin, M.; Hotta, H.; Juniastuti Soetjipto Hayashi, Y.; Hotta, H. Complete Genome Sequence and Phylogenetic Relatedness of Hepatitis B Virus Isolates in Papua, Indonesia. J. Clin. Microbiol. 2009, 47, 1842–1847. [Google Scholar] [CrossRef] [Green Version]
- Meldal, B.; Moula, N.M.; Barnes, I.H.A.; Boukef, K.; Allain, J.-P. A novel hepatitis B virus subgenotype, D7, in Tunisian blood donors. J. Gen. Virol. 2009, 90, 1622–1628. [Google Scholar] [CrossRef]
- Chekaraou, M.A.; Brichler, S.; Mansour, W.; Le Gal, F.; Garba, A.; Dény, P.; Gordien, E. A novel hepatitis B virus (HBV) subgenotype D (D8) strain, resulting from recombination between genotypes D and E, is circulating in Niger along with HBV/E strains. J. Gen. Virol. 2010, 91, 1609–1620. [Google Scholar] [CrossRef]
- Ghosh, S.; Banerjee, P.; Deny, P.; Mondal, R.K.; Nandi, M.; Roychoudhury, A.; Das, K.; Santra, A.; Zoulim, F.; Chowdhury, A.; et al. New HBV subgenotype D9, a novel D/C recombinant, identified in patients with chronic HBeAg-negative infection in Eastern India. J. Viral Hepat. 2012, 20, 209–218. [Google Scholar] [CrossRef]
- Hundie, G.B.; Raj, V.S.; Michael, D.G.; Pas, S.D.; Koopmans, M.P.; Osterhaus, A.D.M.E.; Smits, S.L.; Haagmans, B.L. A novel hepatitis B virus subgenotype D10 circulating in Ethiopia. J. Viral Hepat. 2017, 24, 163–173. [Google Scholar] [CrossRef]
- Ren, C.-C.; Chen, Q.-Y.; Wang, X.-Y.; Harrison, T.J.; Yang, Q.-L.; Hu, L.-P.; Liu, H.-B.; He, X.; Jia, H.-H.; Fang, Z.-L. Novel subgenotype D11 of hepatitis B virus in NaPo County, Guangxi, bordering Vietnam. J. Gen. Virol. 2019, 100, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Lemey, P.; Maes, P.; Van Ranst, M. Are hepatitis B virus “subgenotypes” defined accurately? J. Clin. Virol. 2010, 47, 356–360. [Google Scholar] [CrossRef]
- Schaefer, S.; Magnius, L.; Norder, H. Under Construction: Classification of Hepatitis B Virus Genotypes and Subgenotypes. Intervirology 2009, 52, 323–325. [Google Scholar] [CrossRef]
- Yin, Y.; He, K.; Wu, B.; Xu, M.; Du, L.; Liu, W.; Liao, P.; Liu, Y.; He, M. A systematic genotype and subgenotype re-ranking of hepatitis B virus under a novel classification standard. Heliyon 2019, 5, e02556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.; Yu, M.-L.; Dai, C.-Y.; Lin, I.-L.; Yeh, M.-L.; Chuang, W.-L.; Abe, K. Novel quasi-subgenotype D2 of hepatitis B virus identified in Taiwanese aborigines. Virus Genes 2014, 49, 30–37. [Google Scholar] [CrossRef]
- Thijssen, M.; Trovão, N.S.; Mina, T.; Maes, P.; Pourkarim, M.R. Novel hepatitis B virus subgenotype A8 and quasi-subgenotype D12 in African–Belgian chronic carriers. Int. J. Infect. Dis. 2020, 93, 98–101. [Google Scholar] [CrossRef]
- Schneider, A.D.B.; Osiowy, C.; Hostager, R.; Krarup, H.; Børresen, M.; Tanaka, Y.; Morriseau, T.; Wertheim, J.O. Analysis of Hepatitis B Virus Genotype D in Greenland Suggests the Presence of a Novel Quasi-Subgenotype. Front. Microbiol. 2021, 11, 602296. [Google Scholar] [CrossRef]
- Ozaras, R.; Balkan, I.I.; Yemisen, M.; Tabak, F. Epidemiology of HBV subgenotypes D. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 28–37. [Google Scholar] [CrossRef]
- Locarnini, S.A.; Littlejohn, M.; Yuen, L.K.W. Origins and Evolution of the Primate Hepatitis B Virus. Front. Microbiol. 2021, 12, 653684. [Google Scholar] [CrossRef]
- Paraskevis, D.; Angelis, K.; Magiorkinis, G.; Kostaki, E.; Ho, S.Y.; Hatzakis, A. Dating the origin of hepatitis B virus reveals higher substitution rate and adaptation on the branch leading to F/H genotypes. Mol. Phylogenet. Evol. 2015, 93, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Rasche, A.; Sander, A.-L.; Corman, V.M.; Drexler, J.F. Evolutionary biology of human hepatitis viruses. J. Hepatol. 2019, 70, 501–520. [Google Scholar] [CrossRef] [Green Version]
- Bar-Gal, G.K.; Kim, M.J.; Klein, A.; Shin, D.H.; Oh, C.S.; Kim, J.W.; Kim, T.-H.; Kim, S.B.; Grant, P.R.; Pappo, O.; et al. Tracing hepatitis B virus to the 16th century in a Korean mummy. Hepatology 2012, 56, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Ross, Z.P.; Klunk, J.; Fornaciari, G.; Giuffra, V.; Duchêne, S.; Duggan, A.T.; Poinar, D.; Douglas, M.W.; Eden, J.-S.; Holmes, E.C.; et al. The paradox of HBV evolution as revealed from a 16th century mummy. PLoS Pathog. 2018, 14, e1006750. [Google Scholar] [CrossRef] [Green Version]
- Zehender, G.; Ebranati, E.; Gabanelli, E.; Sorrentino, C.; Presti, A.L.; Tanzi, E.; Ciccozzi, M.; Galli, M. Enigmatic origin of hepatitis B virus: An ancient travelling companion or a recent encounter? World J. Gastroenterol. 2014, 20, 7622–7634. [Google Scholar] [CrossRef]
- Zhou, Y.; Holmes, E.C. Bayesian Estimates of the Evolutionary Rate and Age of Hepatitis B Virus. J. Mol. Evol. 2007, 65, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Spitz, N.; Mello, F.C.A.; Moreira, A.S.; Gusatti, C.S.; Martins, R.M.B.; Gomes, S.A.; Bello, G.; Araujo, N.M. Reconstruction of the spatial and temporal dynamics of hepatitis B virus genotype D in the Americas. PLoS ONE 2019, 14, e0220342. [Google Scholar] [CrossRef]
- Ciccozzi, M.; Ciccaglione, A.R.; Presti, A.L.; Equestre, M.; Cella, E.; Ebranati, E.; Gabanelli, E.; Villano, U.; Bruni, R.; Yalcinkaya, T.; et al. Evolutionary dynamics of HBV-D1 genotype epidemic in Turkey. J. Med. Virol. 2014, 86, 109–116. [Google Scholar] [CrossRef]
- Michitaka, K.; Tanaka, Y.; Horiike, N.; Duong, T.N.; Chen, Y.; Matsuura, K.; Hiasa, Y.; Mizokami, M.; Onji, M. Tracing the history of hepatitis B virus genotype D in western Japan. J. Med. Virol. 2006, 78, 44–52. [Google Scholar] [CrossRef]
- Paraskevis, D.; Magiorkinis, G.; Magiorkinis, E.; Ho, S.Y.; Belshaw, R.; Allain, J.-P.; Hatzakis, A. Dating the origin and dispersal of hepatitis B virus infection in humans and primates. Hepatology 2013, 57, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Trovão, N.S.; Thijssen, M.; Vrancken, B.; Pineda-Peña, A.-C.; Mina, T.; Amini-Bavil-Olyaee, S.; Lemey, P.; Baele, G.; Pourkarim, M.R. Reconstruction of the origin and dispersal of the worldwide dominant Hepatitis B Virus subgenotype D1. Virus Evol. 2022, 8, veac028. [Google Scholar] [CrossRef]
- Zehender, G.; Ebranati, E.; Gabanelli, E.; Shkjezi, R.; Lai, A.; Sorrentino, C.; Presti, A.L.; Basho, M.; Bruno, R.; Tanzi, E.; et al. Spatial and Temporal Dynamics of Hepatitis B Virus D Genotype in Europe and the Mediterranean Basin. PLoS ONE 2012, 7, e37198. [Google Scholar] [CrossRef] [Green Version]
- Zehender, G.; Shkjezi, R.; Ebranati, E.; Gabanelli, E.; Abazaj, Z.; Tanzi, E.; Kraja, D.; Bino, S.; Ciccozzi, M.; Galli, M. Reconstruction of the epidemic history of hepatitis B virus genotype D in Albania. Infect. Genet. Evol. 2012, 12, 291–298. [Google Scholar] [CrossRef]
- Marcelino, R.; Ezeonwumelu, I.J.; Janeiro, A.; Mimoso, P.; Matos, S.; Briz, V.; Pimentel, V.; Pingarilho, M.; Marinho, R.T.; Marcelino, J.M.; et al. Phylogeography of hepatitis B virus: The role of Portugal in the early dissemination of HBV worldwide. PLoS ONE 2022, 17, e0276618. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Banerjee, P.; RoyChoudhury, A.; Sarkar, S.; Ghosh, A.; Santra, A.; Banerjee, S.; Das, K.; Dwibedi, B.; Kar, S.K.; et al. Unique Hepatitis B Virus Subgenotype in a Primitive Tribal Community in Eastern India. J. Clin. Microbiol. 2010, 48, 4063–4071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velkov, S.; Protzer, U.; Michler, T. Global Occurrence of Clinically Relevant Hepatitis B Virus Variants as Found by Analysis of Publicly Available Sequencing Data. Viruses 2020, 12, 1344. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tapias, J.M.; Costa, J.; Mas, A.; Bruguera, M.; Rodés, J. Influence of hepatitis B virus genotype on the long-term outcome of chronic hepatitis B in western patients. Gastroenterology 2002, 123, 1848–1856. [Google Scholar] [CrossRef]
- Sumi, H.; Yokosuka, O.; Seki, N.; Arai, M.; Imazeki, F.; Kurihara, T.; Kanda, T.; Fukai, K.; Kato, M.; Saisho, H. Influence of hepatitis B virus genotypes on the progression of chronic type B liver disease. Hepatology 2003, 37, 19–26. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. Associations Between Hepatitis B Virus Mutations and the Risk of Hepatocellular Carcinoma: A Meta-Analysis. J. Natl. Cancer Inst. 2009, 101, 1066–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, P.K.; Biswas, A.; Datta, S.; Banerjee, A.; Panigrahi, R.; Chakrabarti, S.; De, B.K.; Chakravarty, R. Subgenotypes of hepatitis B virus genotype D (D1, D2, D3 and D5) in India: Differential pattern of mutations, liver injury and occult HBV infection. J. Viral Hepat. 2009, 16, 749–756. [Google Scholar] [CrossRef]
- Datta, S.; Dasgupta, D.; Ghosh, A.; Ghosh, S.; Manna, A.; Datta, S.; Chatterjee, M.; Chowdhury, A.; Banerjee, S. Oncogenic potential of hepatitis B virus subgenotype D1 surpasses D3: Significance in the development of hepatocellular carcinoma. Carcinogenesis 2018, 39, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Khatun, M.; Mondal, R.K.; Pal, S.; Baidya, A.; Bishnu, D.; Banerjee, P.; Santra, A.K.; Dhali, G.K.; Banerjee, S.; Chowdhury, A.; et al. Distinctiveness in virological features and pathogenic potentials of subgenotypes D1, D2, D3 and D5 of Hepatitis B virus. Sci. Rep. 2018, 8, 80551. [Google Scholar] [CrossRef] [Green Version]
- Buster, E.H.; Hansen, B.; Lau, G.K.; Piratvisuth, T.; Zeuzem, S.; Steyerberg, E.; Janssen, H.L. Factors That Predict Response of Patients with Hepatitis B e Antigen–Positive Chronic Hepatitis B to Peginterferon-Alfa. Gastroenterology 2009, 137, 2002–2009. [Google Scholar] [CrossRef]
- Janssen, L.H.; van Zonneveld, M.; Senturk, H.; Zeuzem, S.; Akarca, U.S.; Cakaloglu, Y.; Simon, C.; So, T.M.; Gerken, G.; de Man, R.A.; et al. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: A randomised trial. Lancet 2005, 365, 123–129. [Google Scholar] [CrossRef]
- Wiegand, J.; Hasenclever, D.; Tillmann, H.L. Should Treatment of Hepatitis B Depend on Hepatitis B virus Genotypes? A Hypothesis Generated from an Explorative Analysis of Published Evidence. Antivir. Ther. 2008, 13, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, T.; Suzuki, F.; Kobayashi, M.; Seko, Y.; Kawamura, Y.; Sezaki, H.; Akuta, N.; Suzuki, Y.; Saitoh, S.; Arase, Y.; et al. Clearance of hepatitis B surface antigen during long-term nucleot(s)ide analog treatment in chronic hepatitis B: Results from a nine-year longitudinal study. J. Gastroenterol. 2013, 48, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Sarin, S.K.; Rehman, S.; Guptan, R.C.; Kazim, S.N.; Kumar, S. Role of HBV genotype in predicting response to lamivudine therapy in patients with chronic hepatitis B. Indian J. Gastroenterol. 2005, 24, 12–15. [Google Scholar] [PubMed]
- Khatun, M.; Kumar, K.; Baidya, A.; Mondal, R.K.; Baszczyňski, O.; Kalčic, F.; Banerjee, S.; Dhali, G.K.; Das, K.; Chowdhury, A.; et al. Variability in the Responses of Hepatitis B Virus D-Subgenotypes to Antiviral Therapy: Designing Pan-D-Subgenotypic Reverse Transcriptase Inhibitors. J. Virol. 2022, 96, e0180021. [Google Scholar] [CrossRef]
- Zuckerman, J.N.; Zuckerman, A.J. Mutations of the surface protein of hepatitis B virus. Antivir. Res. 2003, 60, 75–78. [Google Scholar] [CrossRef]
- Carman, W.F.; Zanetti, A.R.; Karayiannis, P.; Waters, J.; Manzillo, G.; Tanzi, E.; Zuckerman, A.J.; Thomas, H.C. Vaccine-induced escape mutant of hepatitis B virus. Lancet 1990, 336, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-H.; Yuan, Q.; Chen, P.-J.; Zhang, Y.-L.; Chen, C.-R.; Zheng, Q.-B.; Yeh, S.-H.; Yu, H.; Xue, Y.; Chen, Y.-X.; et al. Influence of mutations in hepatitis B virus surface protein on viral antigenicity and phenotype in occult HBV strains from blood donors. J. Hepatol. 2012, 57, 720–729. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, Y. Comprehensive analysis of the prevalence of hepatitis B virus escape mutations in the major hydrophilic region of surface antigen. J. Med. Virol. 2011, 84, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Di Lello, F.A.; Ridruejo, E.; Martínez, A.P.; Pérez, P.S.; Campos, R.H.; Flichman, D.M. Molecular epidemiology of hepatitis B virus mutants associated with vaccine escape, drug resistance and diagnosis failure. J. Viral Hepat. 2019, 26, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Elizalde, M.M.; Tadey, L.; Mammana, L.; Quarleri, J.F.; Campos, R.H.; Flichman, D.M. Biological Characterization of Hepatitis B virus Genotypes: Their Role in Viral Replication and Antigen Expression. Front. Microbiol. 2021, 12, 758613. [Google Scholar] [CrossRef]
- Sugiyama, M.; Tanaka, Y.; Kato, T.; Orito, E.; Ito, K.; Acharya, S.K.; Gish, R.G.; Kramvis, A.; Shimada, T.; Izumi, N.; et al. Influence of hepatitis B virus genotypes on the intra- and extracellular expression of viral DNA and antigens. Hepatology 2006, 44, 915–924. [Google Scholar] [CrossRef]
- Zhang, F.; Tang, X.; Garcia, T.; Lok, A.S.; Wang, Y.; Jia, H.; Qin, Y.; Chen, C.; Wen, Y.; Li, J.; et al. Characterization of contrasting features between hepatitis B virus genotype A and genotype D in small envelope protein expression and surface antigen secretion. Virology 2017, 503, 52–61. [Google Scholar] [CrossRef]
- Riveiro-Barciela, M.; Bes, M.; Rodríguez-Frías, F.; Tabernero, D.; Ruiz, A.; Casillas, R.; Vidal-González, J.; Homs, M.; Nieto, L.; Sauleda, S.; et al. Serum hepatitis B core-related antigen is more accurate than hepatitis B surface antigen to identify inactive carriers, regardless of hepatitis B virus genotype. Clin. Microbiol. Infect. 2017, 23, 860–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salpini, R.; Battisti, A.; Piermatteo, L.; Carioti, L.; Anastasiou, O.E.; Gill, U.S.; Di Carlo, D.; Colagrossi, L.; Duca, L.; Bertoli, A.; et al. Key mutations in the C-terminus of the HBV surface glycoprotein correlate with lower HBsAg levels in vivo, hinder HBsAg secretion in vitro and reduce HBsAg structural stability in the setting of HBeAg-negative chronic HBV genotype-D infection. Emerg. Microbes Infect. 2020, 9, 928–939. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Panda, S.K.; Acharya, S.K.; Durgapal, H. Role of hepatitis B virus genotype D & its mutants in occult hepatitis B infection. Indian J. Med. Res. 2013, 138, 329–339. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Genotype D (n = 1156) 1 | ||
|---|---|---|
| Subgenotype (%) | Country (%) | Region (%) 2 |
| D1 (44.3) | Iran (25.2) India (17.6) Turkey (11.5) China (6.6) New Zealand (6.6) Syria (5.9) Lebanon (5.3) Russia (4.9) Tunisia (2.7) Belgium (2.1) Argentina (1.8) Mongolia (1.2) Italy (1.0) Pakistan (1.0) Uzbekistan (1.0) Egypt (0.8) South Africa (0.6) USA (0.6) Belarus (0.4) Greece (0.4) Japan (0.4) Poland (0.4) Bangladesh (0.2) Brazil (0.2) Cuba (0.2) France (0.2) Germany (0.2) Kazakhstan (0.2) Latvia (0.2) Serbia (0.2) Spain (0.2) Tajikistan (0.2) Venezuela (0.2) | Southern Asia (43.9) Western Asia (22.7) Eastern Asia (8.2) Australia and New Zealand (6.6) Eastern Europe (5.7) Northern Africa (3.5) Western Europe (2.5) South America (2.1) Southern Europe (1.8) Central Asia (1.4) Northern America (0.6) Southern Africa (0.6) Caribbean (0.2) Northern Europe (0.2) |
| D2 (24.2) | USA (25) Russia (16.8) India (16.4) Japan (9.3) Brazil (3.6) Estonia (3.6) Belgium (2.5) Spain (2.5) Bangladesh (2.1) Latvia (1.8) Lebanon (1.8) New Caledonia (1.8) Poland (1.8) Argentina (1.1) Belarus (1.1) Ethiopia (1.1) Iran (1.1) Malaysia (1.1) Germany (0.7) Greece (0.7) Tajikistan (0.7) Turkey (0.7) Sudan (0.7) Cuba (0.4) Greenland (0.4) Kazakhstan (0.4) Serbia (0.4) Sweden (0.4) Syria (0.4) | Northern America (25.4) Southern Asia (19.6) Eastern Europe (19.6) Eastern Asia (9.3) Northern Europe (5.7) South America (4.6) Southern Europe (3.6) Western Europe (3.2) Western Asia (2.9) Melanesia (1.8) Central Asia (1.1) Eastern Africa (1.1) Southeastern Asia (1.1) Northern Africa (0.7) Caribbean (0.4) |
| D3 + D6 (16.9) | India (34.4) Brazil (16.4) Italy (13.8) Argentina (6.2) Russia (3.6) Botswana (3.1) China (3.1) South Africa (3.1) Sweden (2.6) Belgium (2.1) Belarus (1.5) Canada (1.5) Serbia (1.5) France (1.0) Haiti (1.0) Sudan (1.0) Cuba (0.5) Estonia (0.5) Germany (0.5) Kazakhstan (0.5) Mongolia (0.5) Spain (0.5) Syria (0.5) USA (0.5) | Southern Asia (34.4) South America (22.6) Southern Europe (15.9) Southern Africa (6.2) Eastern Europe (5.1) Eastern Asia (3.6) Western Europe (3.6) Northern Europe (3.1) Northern America (2.1) Caribbean (1.5) Northern Africa (1.0) Central Asia (0.5) Western Asia (0.5) |
| D4 (1.8) | Brazil (28.6) India (19.0) Cuba (14.3) Canada (9.5) Haiti (9.5) Australia (4.8) New Zealand (4.8) South Africa (4.8) Spain (4.8) | South America (28.6) Caribbean (23.8) Southern Asia (19.0) Australia and New Zealand (9.5) Northern America (9.5) Southern Africa (4.8) Southern Europe (4.8) |
| D5 (1.8) | India (95.2) Bangladesh (4.8) | Southern Asia (100) |
| D7 (2.9) | Tunisia (100) | Northern Africa (100) |
| D8 (0.3) | Niger (100) | Western Africa (100) |
| D9 (0.5) | India (100) | Southern Asia (100) |
| D10 (1.3) | Ethiopia (100) | Eastern Africa (100) |
| D11 (2.0) | China (100) | Eastern Asia (100) |
| quasi-subgenotype D12 (0.2) | Belgium (100) | Western Europe (100) |
| quasi-subgenotype D2 (0.3) | China (100) | Eastern Asia (100) |
| quasi-subgenotype D (3.4) | Greenland (100) | Northern America (100) |
| HBV/D Subgenotype Association | Reference |
|---|---|
| D1 has a significantly higher rate of HCC-associated mutations than D2–D6. | [14] |
| D1 is more significantly associated with chronic liver disease with an elevated ALT compared with D2, D3, and D5. | [61] |
| D1 is more significantly associated with a higher HBsAg positivity rate than D2, D3, and D5. | [61] |
| D1 infection is associated with a higher susceptibility to severe liver damage and rapid progression to HCC than D3. | [62] |
| D1 replicates faster and triggers more ER stress than D3 in vitro. | [62] |
| D1 is associated with a higher risk of HCC than D2 and D3. | [63] |
| HBV/D Subgenotype Association | Reference |
|---|---|
| D1/D2 have a greater susceptibility to TDF/ETV than D3/D5 in vitro. | [69] |
| D1/D2 viral loads are lower than D3/D5 in TDF-treated patients. | [69] |
| D1 has a greater susceptibility to ETV than D3 in vitro. | [62] |
| D2 has a significantly higher rate of LAM-resistant mutants than D1–D6. | [14] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sant’Anna, T.B.; Araujo, N.M. Hepatitis B Virus Genotype D: An Overview of Molecular Epidemiology, Evolutionary History, and Clinical Characteristics. Microorganisms 2023, 11, 1101. https://doi.org/10.3390/microorganisms11051101
Sant’Anna TB, Araujo NM. Hepatitis B Virus Genotype D: An Overview of Molecular Epidemiology, Evolutionary History, and Clinical Characteristics. Microorganisms. 2023; 11(5):1101. https://doi.org/10.3390/microorganisms11051101
Chicago/Turabian StyleSant’Anna, Thaís B., and Natalia M. Araujo. 2023. "Hepatitis B Virus Genotype D: An Overview of Molecular Epidemiology, Evolutionary History, and Clinical Characteristics" Microorganisms 11, no. 5: 1101. https://doi.org/10.3390/microorganisms11051101
APA StyleSant’Anna, T. B., & Araujo, N. M. (2023). Hepatitis B Virus Genotype D: An Overview of Molecular Epidemiology, Evolutionary History, and Clinical Characteristics. Microorganisms, 11(5), 1101. https://doi.org/10.3390/microorganisms11051101