
Leishmania and the Model of Predominant Clonal Evolution



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. A Brief Recall on the Clonal Theory in Its Present Form: The Predominant Clonal Evolution Model (PCE)
2.1. What Are the Units of Analysis Considered by the PCE Approach?
2.2. Where to Put the Cursor?
2.3. The “Clonality Threshold”: Phylogenetic Signal/Clonality Backbone/Bifurcating Trees
2.4. The Near-Clade (NC) Concept
2.5. Other PCE Features
2.6. Side Concepts: Clonet, Russian Doll Pattern (RDP)
2.7. Resolution of Markers and the PCE Model
2.8. The Importance of Sampling Strategies
3. Leishmania and the PCE Model
3.1. Widespread Clonal Genotypes
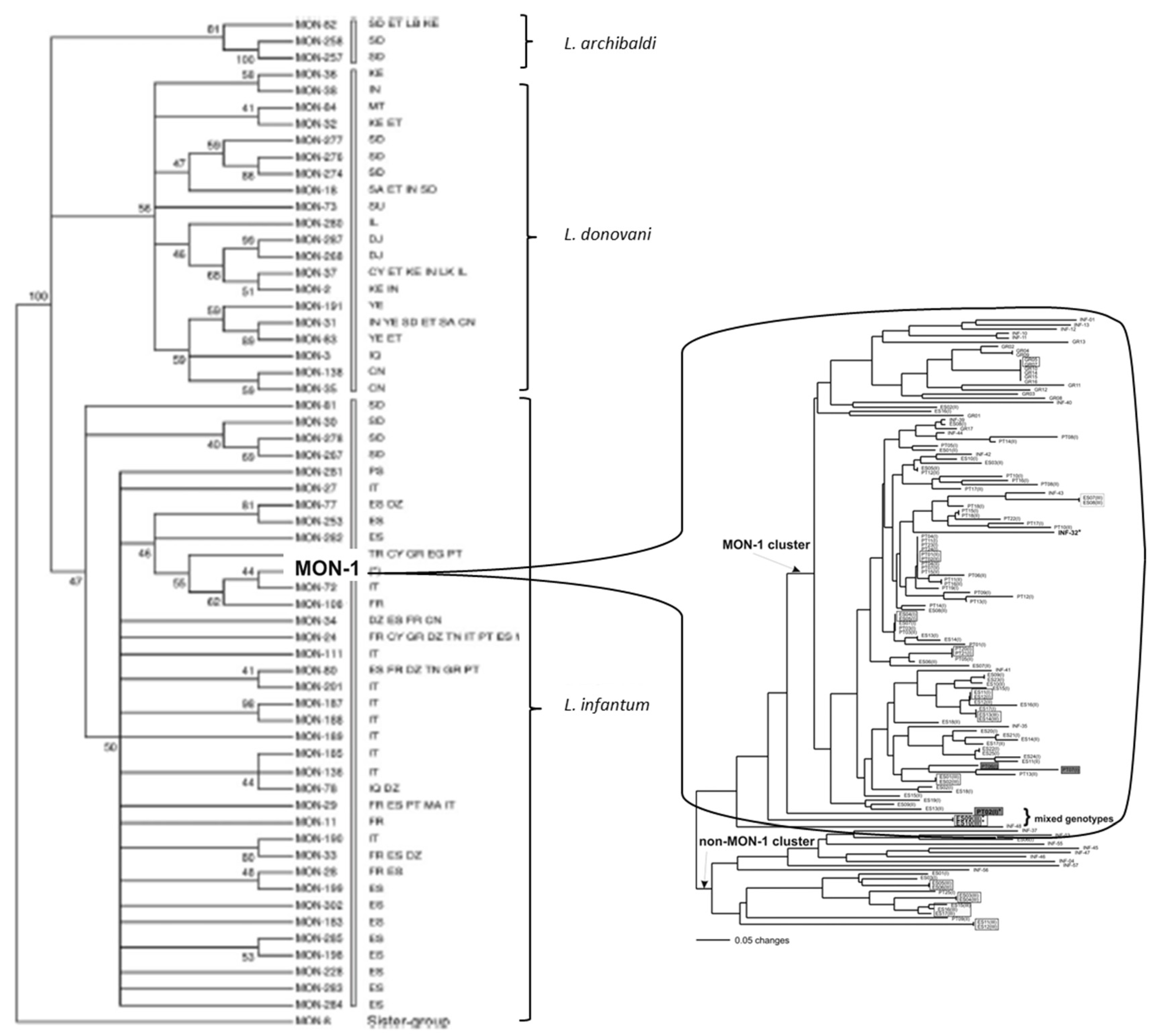
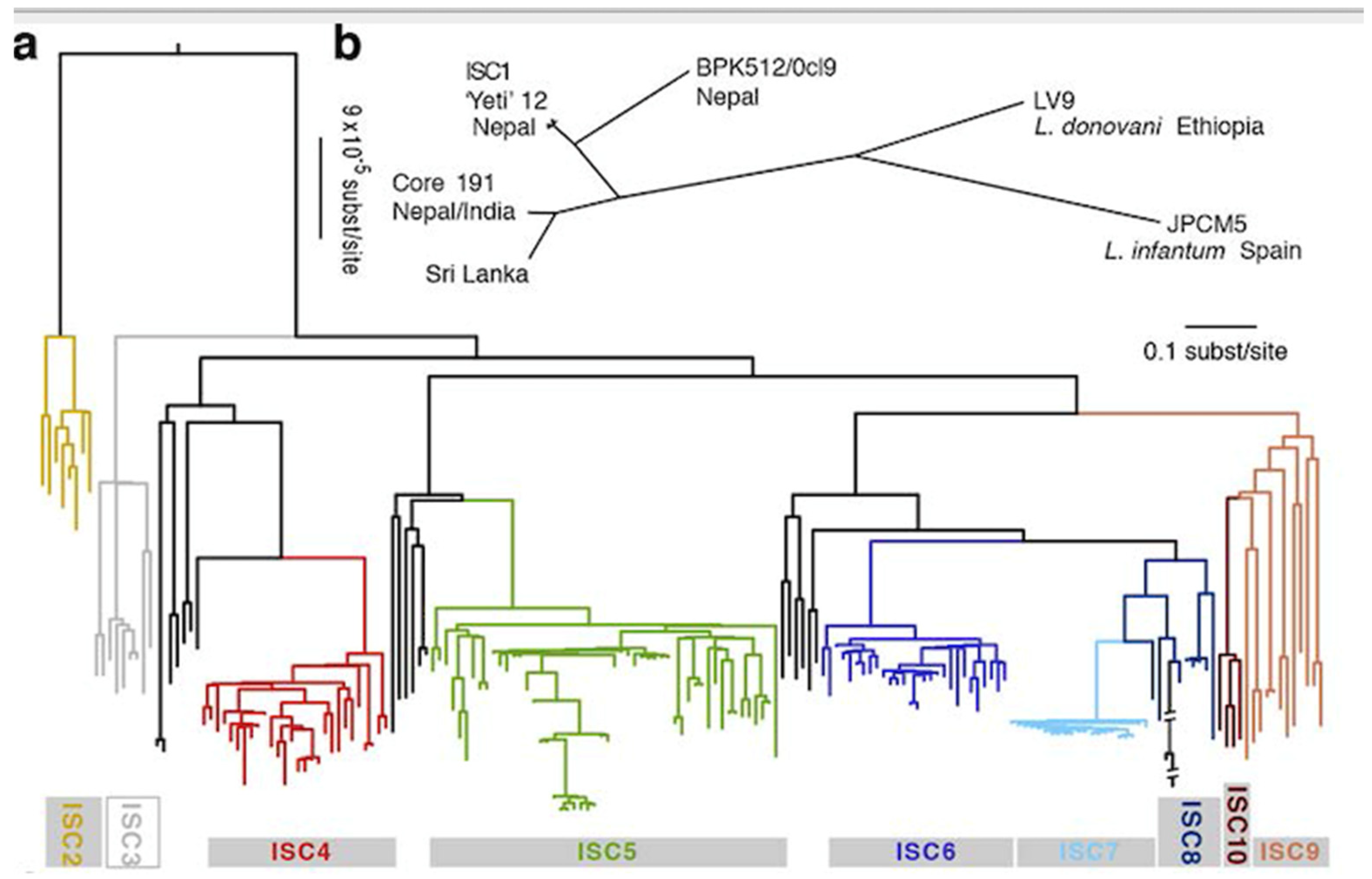
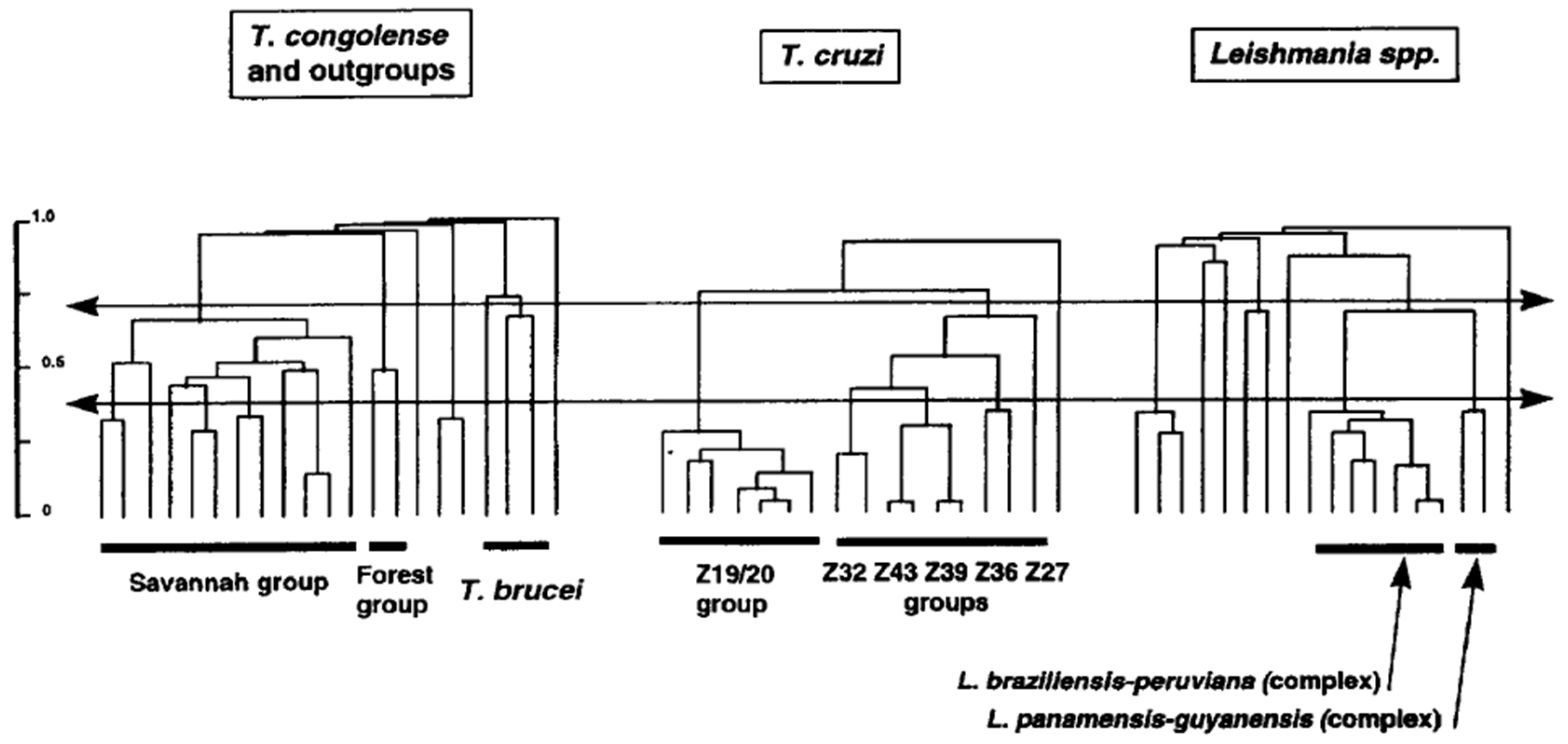
3.2. Deep Phylogenies, Bifurcating Trees, Near-Clades
3.3. Linkage Disequilibrium (LD)
3.4. Within-Species Diversity—Russian Doll Patterns (RDPs)
3.5. Leishmania: Species, or Near-Clades?
3.6. Widespread Aneuploidy, Population Genetics and the PCE Mode
3.7. Experimental Mating, Hybrid Lines, Meiosis Genes, and the PCE Model
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Selander, R.K.; Levin, B.R. Genetic diversity and structure in Escherichia coli populations. Science 1980, 210, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Ørskov, F.; Ørskov, I. Summary of a workshop on the clone concept in the epidemiology, taxonomy, and evolution of the Enterobacteriaceae and other Bacteria. J. Infect. Dis. 1983, 148, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Tibayrenc, M.; Cariou, M.L.; Solignac, M.; Carlier, Y. Arguments génétiques contre l’existence d’une sexualité actuelle chez Trypanosoma cruzi; implications taxinomiques. C. R. Acad. Sci. Paris 1981, 293, 207–209. [Google Scholar]
- Tibayrenc, M.; Kjellberg, F.; Ayala, F.J. A clonal theory of parasitic protozoa: The population structure of Entamoeba, Giardia, Leishmania, Naegleria, Plasmodium, Trichomonas and Trypanosoma, and its medical and taxonomical consequences. Proc. Nat. Acad. Sci. USA 1990, 87, 2414–2418. [Google Scholar] [CrossRef] [Green Version]
- Tibayrenc, M.; Ayala, F.J. Reproductive clonality of pathogens: A perspective on pathogenic viruses, bacteria, fungi, and parasitic protozoa. Proc. Nat. Acad. Sci. USA 2012, 109, E3305–E3313. [Google Scholar] [CrossRef] [Green Version]
- Tibayrenc, M.; Ayala, F.J. Is predominant clonal evolution a common evolutionary adaptation to parasitisms in parasitic protozoa, fungi, bacteria and viruses? Adv. Parasitol. 2017, 96, 243–325. [Google Scholar]
- Tibayrenc, M.; Ayala, F.J. Models in parasite and pathogen evolution: Genomic analysis reveals predominant clonality and progressive evolution at all evolutionary scales in parasitic protozoa, yeasts and bacteria. Adv. Parasitol. 2021, 111, 75–117. [Google Scholar] [PubMed]
- Avise, J.C. Evolutionary perspectives on clonal reproduction in vertebrate animals. Proc. Nat. Acad. Sci. USA 2015, 112, 8867–8873. [Google Scholar] [CrossRef] [Green Version]
- Rougeron, V.; De Meeûs, T.; Hide, M.; Waleckx, E.; Bermudez, H.; Arevalo, J.; Llanos-Cuentas, A.; Dujardin, J.C.; De Doncker, S.; Le Ray, D.; et al. Extreme inbreeding in Leishmania braziliensis. Proc. Nat. Acad. Sci. USA 2009, 106, 10224–10229. [Google Scholar] [CrossRef] [Green Version]
- Hauck, S.; Maiden, M.C. Clonally Evolving Pathogenic Bacteria in: P H Rampelotto (eD). In Molecular Mechanisms of Microbial Evolution; Grand Challenges in Biology and Biotechnology; Springer: New York, NY, USA, 2018; pp. 307–325. [Google Scholar]
- Gutiérrez-Corbo, C.; Domínguez-Asenjo, B.; Martínez-Valladares, M.; Pérez-Pertejo, Y.; García-Estrada, C.; Balaña-Fouce, R.; Reguera, R.M. Reproduction in Trypanosomatids: Past and Present. Biology 2021, 10, 471. [Google Scholar] [CrossRef]
- Rougeron, V.; De Meeûs, T.; Hide, M.; Le Falher, G.; Bucheton, B.; Dereure, J.; El-Safi, S.H.; Dessein, A.; Bañuls, A.L. Multifaceted Population Structure and Reproductive Strategy in Leishmania donovani Complex in One Sudanese Village. PLoS Negl. Trop. Dis. 2011, 5, e1448. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.D.; Llewellyn, M.S.; Yeo, M.; Messenger, L.A.; Miles, M.A. Experimental and natural recombination in Trypanosoma cruzi. In American Trypanosomiasis: Chagas Disease. One Hundred Years of Research, 2nd ed.; Telleria, J., Tibayrenc, M., Eds.; Elsevier/Academic Press: Amsterdam, The Netherlands, 2017; Chapter 20. [Google Scholar]
- Ramírez, J.D.; Llewellyn, J.D. Reproductive clonality in protozoan pathogens—Truth or artefact? Molec. Ecol. 2014, 23, 4195–4202. [Google Scholar] [CrossRef] [PubMed]
- Hanage, W.P. Not So Simple After All: Bacteria, Their Population Genetics, and Recombination. Cold Spring Harb. Perspect. Biol. 2016, 8, a018069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiden, M.C.J. Multilocus Sequence Typing of Bacteria. Ann. Rev. Microbiol. 2006, 60, 561–588. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.; Giffard, P.; Beckstrom-Sternberg, S.; Auerbach, R.; Hornstra, H.; Tuanyok, A.; Price, E.P.; Glass, M.B.; Leadem, B.; Beckstrom-Sternberg, J.S.; et al. Phylogeographic reconstruction of a bacterial species with high levels of lateral gene transfer. BMC Biol. 2009, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Losada, M.; Browne, E.B.; Madsen, A.; Wirth, T.; Viscidi, R.P.; Crandall, K.A. Population genetics of microbial pathogens estimated from multilocus sequence typing (MLST) data. Infect. Genet. Evol. 2006, 6, 97–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.F.; Guttman, D.S. Evolution of the Core Genome of Pseudomonas syringae, a Highly Clonal, Endemic Plant Pathogen. Appl. Environ. Microbiol. 2004, 70, 1999–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Awadalla, P. The evolutionary genomics of pathogen recombination. Nat. Rev. Genet. 2003, 4, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Rougeron, V.; De Meeûs, T.; Kako Ouraga, S.; Hide, M.; Bañuls, A.L. ‘‘Everything You Always Wanted to Know about Sex (but Were Afraid to Ask)’’ in Leishmania after Two Decades of Laboratory and Field Analyses. PLos Pathog. 2010, 6, e1001004. [Google Scholar] [CrossRef]
- Tibayrenc, M.; Kjellberg, F.; Ayala, F.J. The clonal theory of parasitic protozoa: A taxonomic proposal applicable to other clonal organisms. Bioscience 1991, 41, 767–774. [Google Scholar] [CrossRef]
- Tibayrenc, M.; Ayala, F.J. How clonal are Trypanosoma and Leishmania? Trends Parasitol. 2013, 29, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Gelanew, T.; Kuhls, K.; Hurissa, Z.; Weldegebreal, T.; Hailu, W.; Kassahun, A.; Abebe, T.; Hailu, A.; Schönian, G. Inference of Population Structure of Leishmania donovani Strains Isolated from Different Ethiopian Visceral Leishmaniasis Endemic Areas. PLoS Neglect. Trop. Dis. 2010, 4, e889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauricio, I.L.; Yeo, M.; Baghaei, M.; Doto, D.; Pratlong, F.; Zemanova, E.; Dedet, J.P.; Lukes, J.; Miles, M.A. Towards multilocus sequence typing of the Leishmania donovani complex: Resolving genotypes and haplotypes for five polymorphic metabolic enzymes (ASAT, GPI, NH1, NH2, PGD). Int. J. Parasitol. 2006, 36, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Shaik, J.S.; Dobson, D.E.; Sacks, D.L.; Beverley, S.M. Leishmania Sexual Reproductive Strategies as Resolved through Computational Methods Designed for Aneuploid Genomes. Genes 2021, 12, 167. [Google Scholar] [CrossRef]
- Kuhls, K.; Chicharro, C.; Cañavate, C.; Cortes, S.; Campino, L.; Haralambous, C.; Soteriadou, K.; Pratlong, F.; Dedet, J.P.; Mauricio, I.; et al. Differentiation and Gene Flow among European Populations of Leishmania infantum MON-1. PLoS Neglect. Trop. Dis. 2008, 2, e261. [Google Scholar] [CrossRef] [Green Version]
- Pratlong, F.; Lami, P.; Ravel, C.; Balard, Y.; Dereure, J.; Serres, G.; El Baidouri, F.; Dedet, J.P. Geographical distribution and epidemiological features of Old World Leishmania infantum and Leishmania donovani foci, based on the isoenzyme analysis of 2277 strains. Parasitol 2013, 40, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochsenreither, S.; Kuhls, K.; Schaar, M.; Presber, W.; Schönian, G. Multilocus Microsatellite Typing as a New Tool for Discrimination of Leishmania infantum MON-1 Strains. J. Clin. Microbiol. 2006, 44, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, G.E.M.; dos Santos, B.N.; Dorval, M.E.C.; Ramos, T.P.B.; Porrozzi, R.; Peixoto, A.A.; Cupolillo, E. The Genetic Structure of Leishmania infantum Populations in Brazil and Its Possible Association with the Transmission Cycle of Visceral Leishmaniasis. PLoS ONE 2012, 77, e36242. [Google Scholar] [CrossRef] [Green Version]
- Cortes, S.; Maurício, I.L.; Kuhls, K.; Nunes, M.; Lopes, C.; Marcos, M.; Cardoso, L.; Schönian, G.; Campino, L. Genetic diversity evaluation on Portuguese Leishmania infantum strains by multilocus microsatellite typing. Infect. Genet. Evol. 2014, 26, 20–31. [Google Scholar] [CrossRef]
- Kuhls, K.; Zahangir Alam, M.; Cupolillo, E.; Ferreira, G.E.M.; Mauricio, I.L.; Oddone, R.; Feliciangeli, M.D.; Wirth, T.; Miles, M.A.; Schönian, G. Comparative Microsatellite Typing of New World Leishmania infantum Reveals Low Heterogeneity among Populations and Its Recent Old World Origin. PLoS Neglect. Trop. Dis. 2011, 5, e1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banu, S.S.; Meyer, W.; Ferreira-Paim, K.; Wanga, Q.; Kuhls, K.; Cupolillo, E.; Schönian, G.; Lee, R. A novel multilocus sequence typing scheme identifying genetic diversity amongst Leishmania donovani isolates from a genetically homogeneous population in the Indian subcontinent. Int. J. Parasitol. 2019, 49, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Franssen, S.U.; Durrant, C.; Stark, O.; Moser, B.; Downing, T.; Imamura, H.; Dujardin, J.C.; Sanders, M.J.; Mauricio, I.; Miles, M.A.; et al. Global genome diversity of the Leishmania donovani complex. eLife 2020, 9, e51243. [Google Scholar] [CrossRef] [PubMed]
- Rugna, G.; Carra, E.; Bergamini, F.; Calzolari, M.; Salvatore, D.; Corpus, F.; Gennari, W.; Baldelli, R.; Fabbi, M.; Natalini, S.; et al. Multilocus microsatellite typing (MLMT) reveals host-related population structure in Leishmania infantum from northeastern Italy. PLoS Negl. Trop. Dis. 2018, 12, e0006595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, G.; Bruno, F.; Caputo, V.; Fiorella, S.; Sammarco, I.; Lupo, T.; Migliazzo, A.; Vitale, F.; Reale, S. Genetic tools discriminate strains of Leishmania infantum isolated from humans and dogs in Sicily, Italy. PLoS Negl. Trop. Dis. 2020, 14, e0008465. [Google Scholar] [CrossRef] [PubMed]
- Amro, A.; Schönian, G.; Al-Sharabati, M.B.B.; Azmi, K.; Nasereddin, A.; Abdeen, Z.; Schnur, L.F.; Baneth, G.; Jaffe, C.L.; Kuhls, K. Population genetics of Leishmania infantum in Israel and the Palestinian Authority through microsatellite analysis. Microbes Infect. 2009, 11, 484–492. [Google Scholar] [CrossRef]
- Chargui, N.; Amro, A.; Haouas, N.; Schönian, G.; Babba, H.; Schmidt, S.; Ravel, C.; Lefebvre, M.; Bastien, P.; Chaker, E.; et al. Population structure of Tunisian Leishmania infantum and evidence for the existence of hybrids and gene flow between genetically different populations. Int. J. Parasitol. 2009, 39, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Kuhls, K.; Moskalenko, O.; Sukiasyan, A.; Manukyan, D.; Melik-Andreasyan, G.; Atshemyan, L.; Apresyan, H.; Strelkova, M.; Jaeschke, A.; Wieland, R.; et al. Microsatellite based molecular epidemiology of Leishmania infantum from re-emerging foci of visceral leishmaniasis in Armenia and pilot risk assessment by ecological niche modeling. PLoS Negl. Trop. Dis. 2021, 15, e0009288. [Google Scholar] [CrossRef] [PubMed]
- Odiwuor, S.; Veland, N.; Maes, I.; Arévalo, J.; Dujardin, J.C.; Van der Auwera, G. Evolution of the Leishmania braziliensis species complex from amplified fragment length polymorphisms, and clinical implications. Infect. Genet. Evol. 2012, 12, 1994–2002. [Google Scholar] [CrossRef] [PubMed]
- Schwenkenbecher, J.M.; Wirth, T.; Schnur, L.F.; Jaffe, C.L.; Schallig, H.; Al-Jawabreh, A.; Hamarsheh, O.; Azmi, K.; Pratlong, F.; Schönian, G. Microsatellite analysis reveals genetic structure of Leishmania tropica. Int. J. Parasitol. 2006, 36, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Chaara, D.; Ravel, C.; Bañuls, A.L.; Haouas, N.; Lami, P.; Talignani, L.; El Baidouri, F.; Jaouadi, K.; Harrat, Z.; Dedet, J.P.; et al. Evolutionary history of Leishmania killicki (synonymous Leishmania tropica) and taxonomic implications. Parasites Vectors 2015, 8, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, D.G.; Monteiro, G.R.G.; Martins, D.R.A.; Fernandes, M.Z.; Macedo-Silva, V.; Ansaldi, M.; Nascimento, P.R.P.; Kurtz, M.A.; Streit, J.A.; Ximenes, M.F.F.M.; et al. Comparative analyses of whole genome sequences of Leishmania infantum isolates from humans and dogs in northeastern Brazil. Int. J. Parasitol. 2017, 47, 655–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, M.B.; Downing, T.; Smith, B.A.; Imamura, H.; Sanders, M.; Svobodova, M.; Volf, P.; Berriman, M.; Cotton, J.A.; Smith, D.F. Genomic Confirmation of Hybridisation and Recent Inbreeding in a Vector-Isolated Leishmania Population. PLoS Genet. 2014, 10, e1004092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, H.; Downing, T.; Van den Broeck, F.; Sanders, M.J.; Rijal, S.; Sundar, S.; Mannaert, A.; Vanaerschot, M.; Berg, M.; De Muylder, G.; et al. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. eLife 2016, 5, e12613. [Google Scholar] [CrossRef] [Green Version]
- Gouzelou, E.; Haralambous, C.; Amro, A.; Mentis, A.; Pratlong, F.; Dedet, J.P.; Votypka, J.; Volf, P.; Toz, S.O.; Kuhls, K.; et al. Multilocus Microsatellite Typing (MLMT) of Strains from Turkey and Cyprus Reveals a Novel Monophyletic L. donovani Sensu Lato Group. PLoS Neglect. Trop. Dis. 2012, 6, e1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seridi, N.; Amro, A.; Kuhls, K.; Belkaid, M.; Zidane, C.; Al-Jawabreh, A.; Schönian, G. Genetic polymorphism of Algerian Leishmania infantum strains revealed by multilocus microsatellite analysis. Microbes Infect. 2008, 10, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Downing, T.; Imamura, H.; Decuypere, S.; Clark, T.G.; Coombs, G.H.; Cotton, J.A.; Hilley, J.D.; de Doncker, S.; Maes, L.; Mottram, J.C.; et al. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011, 21, 2143–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downing, T.; Stark, O.; Vanaerschot, M.; Imamura, H.; Sanders, M.; Decuypere, S.; de Doncker, S.; Maes, I.; Rijal, S.; Sundar, S.; et al. Genome-wide SNP and microsatellite variation illuminate population-level epidemiology in the Leishmania donovani species complex. Infect. Genet. Evol. 2012, 12, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.Z.; Kuhls, K.; Schweynoch, C.; Sundar, S.; Rijal, S.; Shamsuzzaman, A.K.M.; Raju, B.V.S.; Salotra, P.; Dujardin, J.C.; Schönian, G. Multilocus microsatellite typing (MLMT) reveals genetic homogeneity of Leishmania donovani strains in the Indian subcontinent. Infect. Genet. Evol. 2009, 90, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Biek, R.; Pybus, O.G.; Lloyd-Smith, J.O.; Didelot, X. Measurably evolving pathogens in the genomic era. Trends Ecol. Evol. 2015, 30, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Cracraft, J. Species concept and speciation analysis. In Current Ornithology; Johnson, R.F., Ed.; Plenum Press: New York, NY, USA, 1983; pp. 159–187. [Google Scholar]
- Miles, M.A.; Souza, A.; Povoa, M.; Shaw, J.J.; Lainson, R.; Toyé, P.J. Isozymic heterogeneity of Trypanosoma cruzi in the first autochtonous patients with Chagas’disease in Amazonian Brazil. Nature 1978, 272, 819–821. [Google Scholar] [CrossRef] [PubMed]
- Miles, M.A.; Povoa, M.; De Souza, A.A.; Lainson, R.; Shaw, J.J.; Ketteridge, D.S. Chagas’disease in the Amazon Basin: II. The distribution of Trypanosoma cruzi zymodemes 1 and 3 in Para State, north Brazil. Trans. R. Soc. Trop. Med. Hyg. 1981, 75, 667–674. [Google Scholar] [CrossRef]
- Tibayrenc, M. Genetic epidemiology of parasitic protozoa and other infectious agents: The need for an integrated approach. Int. J. Parasitol. 1998, 28, 85–104. [Google Scholar] [CrossRef]
- Boité, M.C.; Mauricio, I.L.; Miles, M.A.; Cupolillo, E. New Insights on Taxonomy, Phylogeny and Population Genetics of Leishmania (Viannia) Parasites Based on Multilocus Sequence Analysis. PLoS Neglect. Trop. Dis. 2012, 6, e1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimont, P.A.D. Use of DNA reassociation in bacterial classification. Can. J. Microbiol. 1998, 34, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Lachaud, L.; Bourgeois, N.; Kuk, N.; Morelle, C.; Crobu, L.; Merlin, G.; Bastien, P.; Pagès, M.; Sterkers, Y. Constitutive mosaic aneuploidy is a unique genetic feature widespread in the Leishmania genus. Microbes Infect. 2014, 16, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef] [Green Version]
- Inbar, E.; Akopyants, N.S.; Charmoy, M.; Romano, A.; Lawyer, P.; Elnaiem, D.A.; Kauffmann, F.; Barhoumi, M.; Grigg, M.; Owens, K.; et al. The Mating Competence of Geographically Diverse Leishmania major Strains in Their Natural and Unnatural Sand Fly Vectors. PLoS Genet. 2013, 9, e1003672. [Google Scholar] [CrossRef] [PubMed]
- Sterkers, Y.; Lachaud, L.; Crobu, L.; Bastien, P.; Pagès, M. FISH analysis reveals aneuploidy and continual generation of chromosomal mosaicism in Leishmania major. Cellular Microbiol. 2011, 139, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Mannaert, A.; Downing, T.; Imamura, H.; Dujardin, J.C. Adaptive mechanisms in pathogens: Universal aneuploidy in Leishmania. Trends Parasitol. 2012, 28, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, J.D.; Guhl, F.; Messager, L.A.; Lewis, M.D.; Montilla, M.; Cucunúba, Z.; Miles, M.A.; Llewellyn, M.S. Contemporary cryptic sexuality in Trypanosoma cruzi. Mol. Ecol. 2012, 17, 4216–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterkers, Y.; Lachaud, L.; Bourgeois, N.; Crobu, L.; Bastien, P.; Pagès, M. Novel insights into genome plasticity in Eukaryotes: Mosaic aneuploidy in Leishmania. Molec. Microbiol. 2012, 86, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akopyants, N.S.; Kimblin, N.; Secundino, N.; Patrick, R.; Peters, N.; Lawyer, P.; Dobson, D.E.; Beverley, S.M.; Sacks, D.L. Demonstration of genetic exchange during cyclical development of Leishmania in the sand fly vector. Science 2009, 324, 265–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inbar, E.; Shaik, J.; Iantorno, S.A.; Romano, A.; Nzelu, C.O.; Owens, K.; Sanders, M.J.; Dobson, D.; CottonI, J.A.; Grigg, M.E.; et al. Whole genome sequencing of experimental hybrids supports meiosis-like sexual recombination in Leishmania. PLoS Genet. 2019, 15, e1008042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravel, C.; Cortes, S.; Pratlong, F.; Morio, F.; Dedet, J.P.; Campino, L. First report of genetic hybrids between two very divergent Leishmania species: Leishmania infantum and Leishmania major. Int. J. Parasitol. 2006, 36, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Schwabl, P.; Boité, M.C.; Bussotti, G.; Jacobs, A.; Andersson, B.; Moreira, O.; Freitas-Mesquita, A.L.; Meyer-Fernandes, J.R.; Telleria, E.L.; Traub-Csekö, Y.; et al. Colonization and genetic diversification processes of Leishmania infantum in the Americas. Nat. Commun. 2021, 4, 139. Available online: www.nature.com/commsbio (accessed on 15 October 2019). [CrossRef]
- Van den Broeck, F.; Savill, N.J.; Imamura, H.; Sanders, M.; Maes, I.; Cooper, S.; Mateus, D.; Jara, M.; Adaui, V.; Arevalo, J.; et al. Ecological divergence and hybridization of Neotropical Leishmania parasites. Proc. Nat. Acad. Sci. USA 2020, 117, 25159–25168. [Google Scholar] [CrossRef]
- Brisse, S.; Henriksson, J.; Barnabé, C.; Douzery, E.J.P.; Berkvens, D.; Serrano, M.; De Carvalho, M.R.C.; Buck, G.A.; Dujardin, J.C.; Tibayrenc, M. Evidence for genetic exchange and hybridization in Trypanosoma cruzi based on nucleotide sequences and molecular karyotype. Infect. Genet. Evol. 2003, 2, 173–183. [Google Scholar] [CrossRef]
- Schwabl, P.; Imamura, H.; Van den Broeck, F.; Costales, J.A.; Maiguashca-Sánchez, J.; Miles, M.A.; Andersson, B.; Grijalva, M.J.; Llewellyn, M.S. Meiotic sex in Chagas disease parasite Trypanosoma cruzi. Nat. Com. 2019, 10, 3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, W.C. The sexual side of parasitic protists. Mol. Biochem. Parasitol. 2021, 243, 371. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W. Sex: Is Giardia doing it in the dark? Curr. Biol. 2005, 15, R56–R58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birky, C.W. Giardia Sex? Yes, but how and how much? Trends Parasitol. 2009, 26, 70–74. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tibayrenc, M.; Ayala, F.J. Leishmania and the Model of Predominant Clonal Evolution. Microorganisms 2021, 9, 2409. https://doi.org/10.3390/microorganisms9112409
Tibayrenc M, Ayala FJ. Leishmania and the Model of Predominant Clonal Evolution. Microorganisms. 2021; 9(11):2409. https://doi.org/10.3390/microorganisms9112409
Chicago/Turabian StyleTibayrenc, Michel, and Francisco J. Ayala. 2021. "Leishmania and the Model of Predominant Clonal Evolution" Microorganisms 9, no. 11: 2409. https://doi.org/10.3390/microorganisms9112409
APA StyleTibayrenc, M., & Ayala, F. J. (2021). Leishmania and the Model of Predominant Clonal Evolution. Microorganisms, 9(11), 2409. https://doi.org/10.3390/microorganisms9112409