
Unraveling the Gut Microbiome of the Invasive Small Indian Mongoose (Urva auropunctata) in the Caribbean



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Trapping Location
2.2. DNA Extraction, PCR, Library Preparation, and Sequencing
2.3. Sequence Processing, Quality Control, OTU Clustering, and Species Annotation
2.4. Data Analyses
3. Results
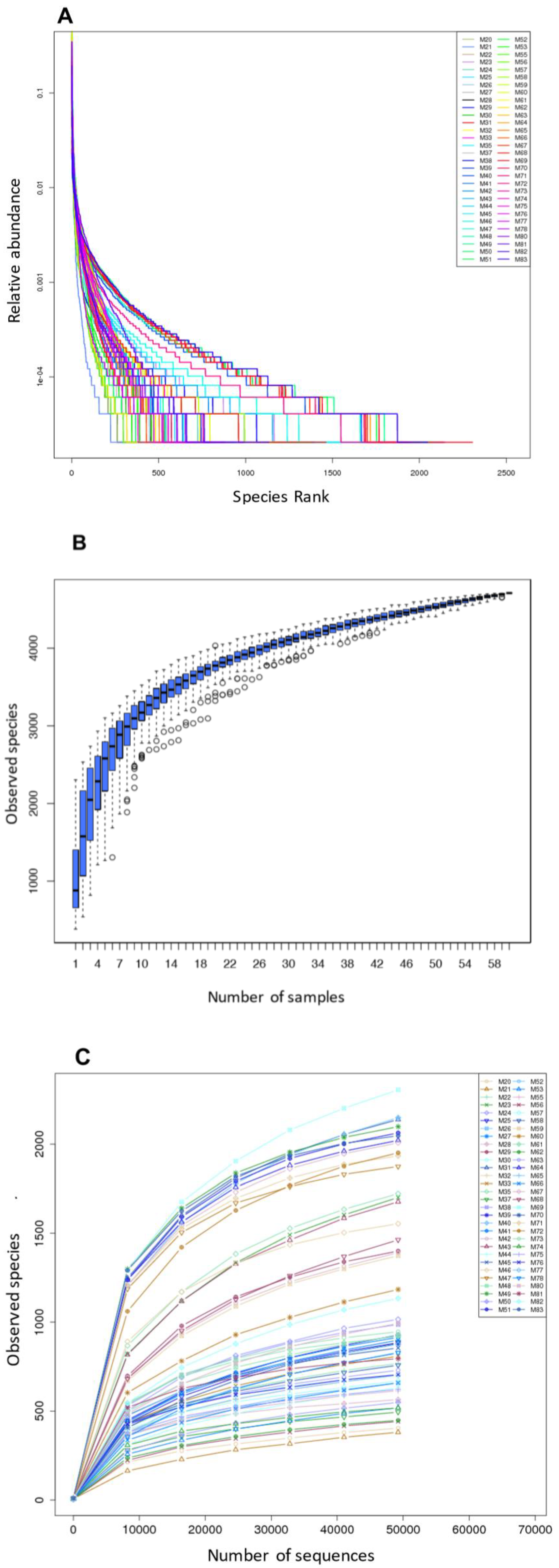
3.1. Sample Collection, Sequencing, and Quality Control
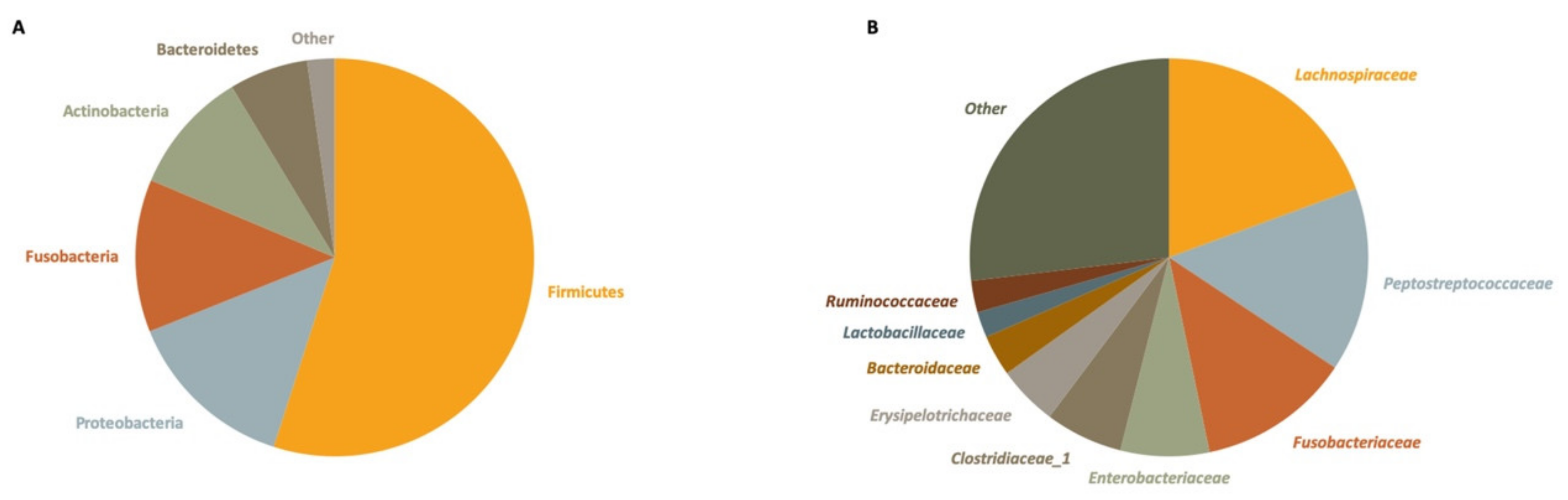
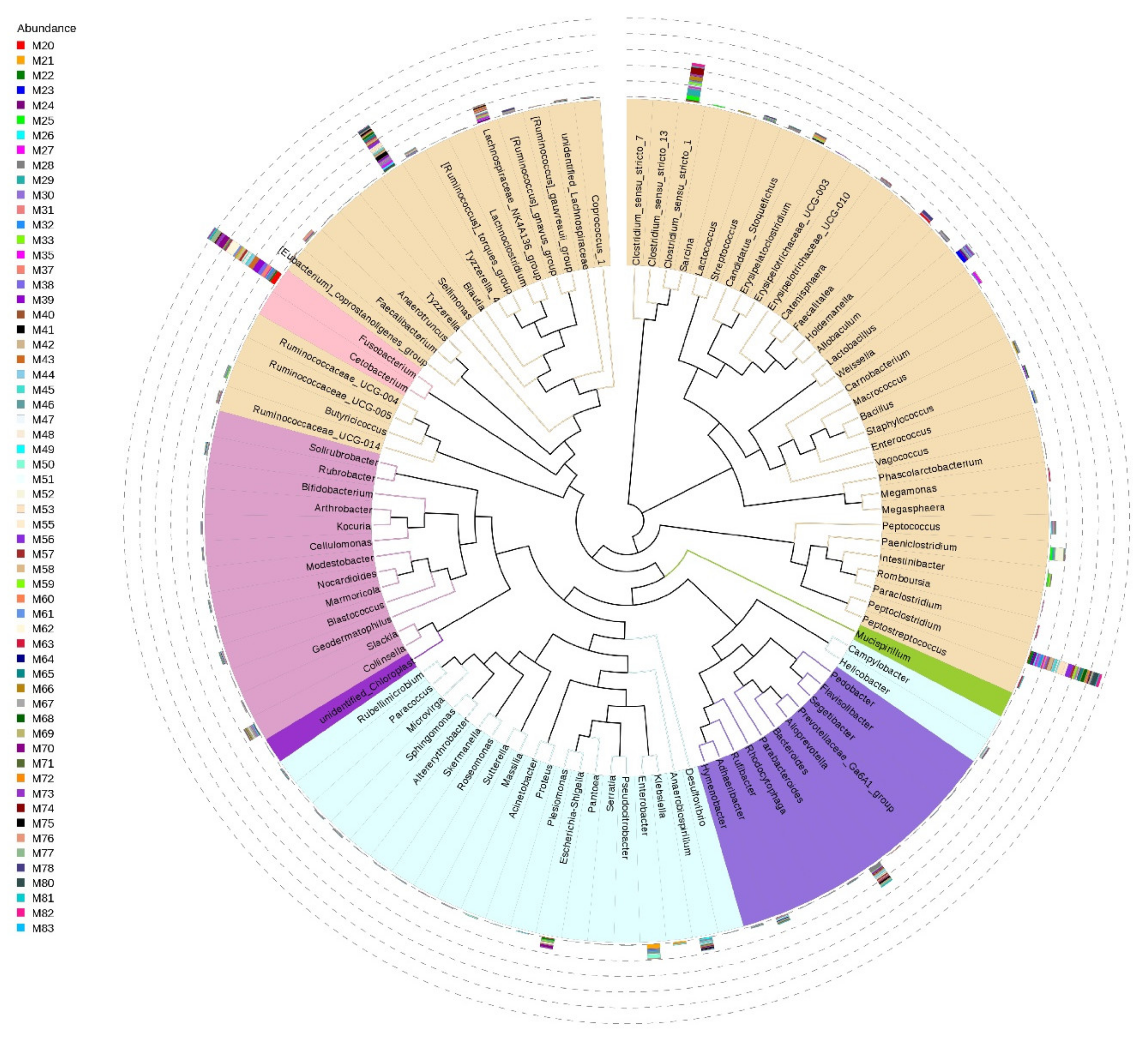
3.2. Gut Microbiota Profile of Small Indian Mongooses (Urva auropunctata)
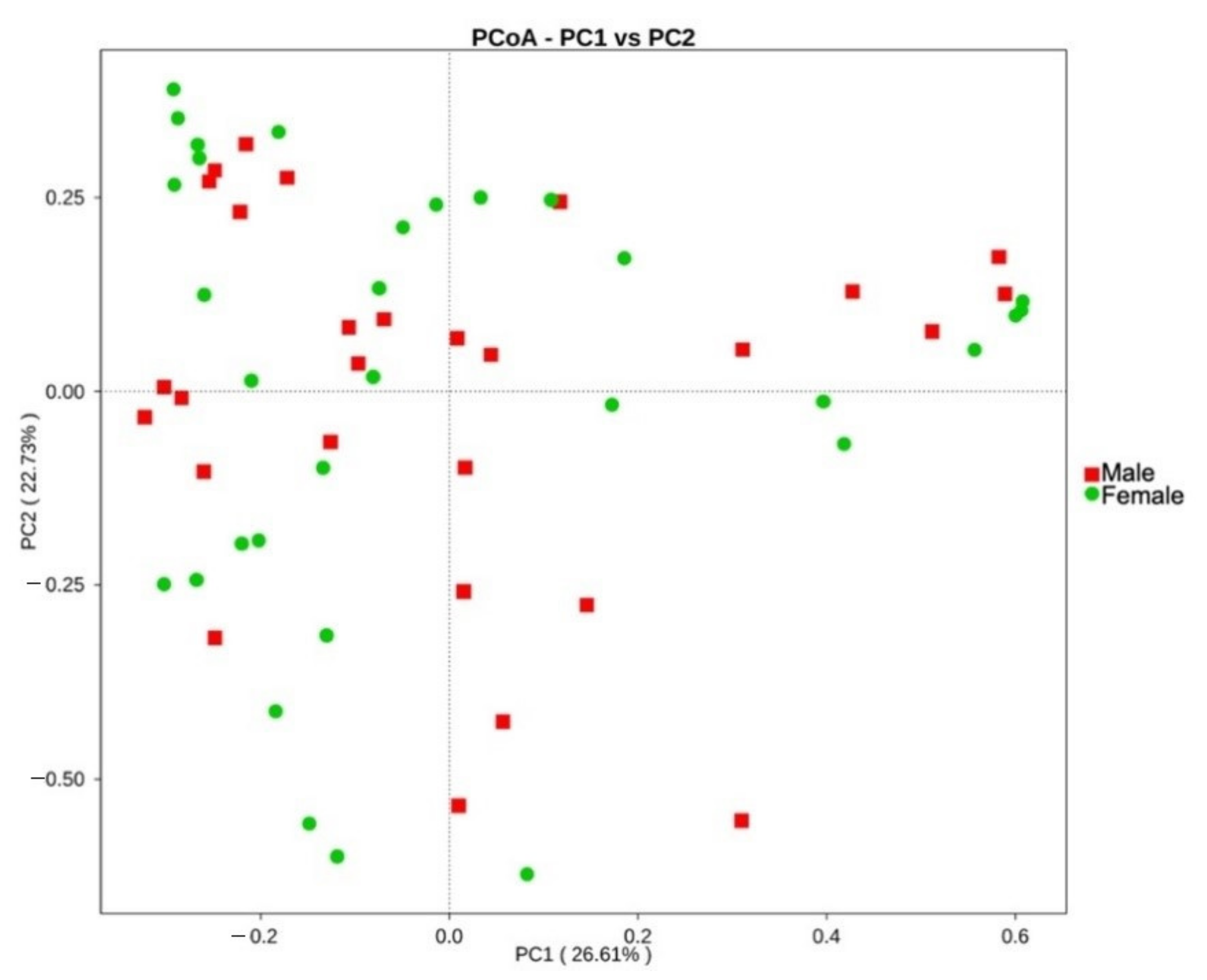
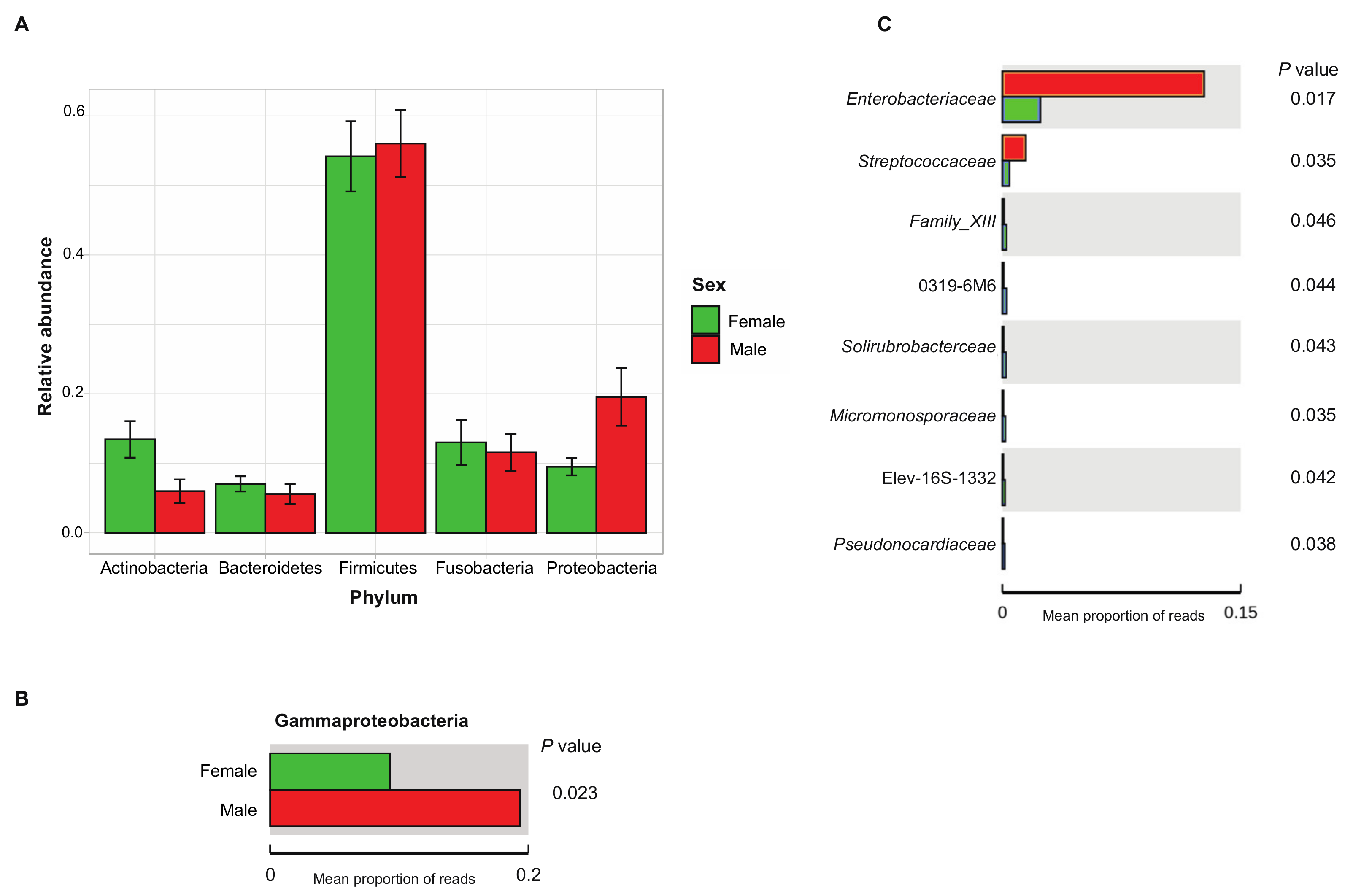
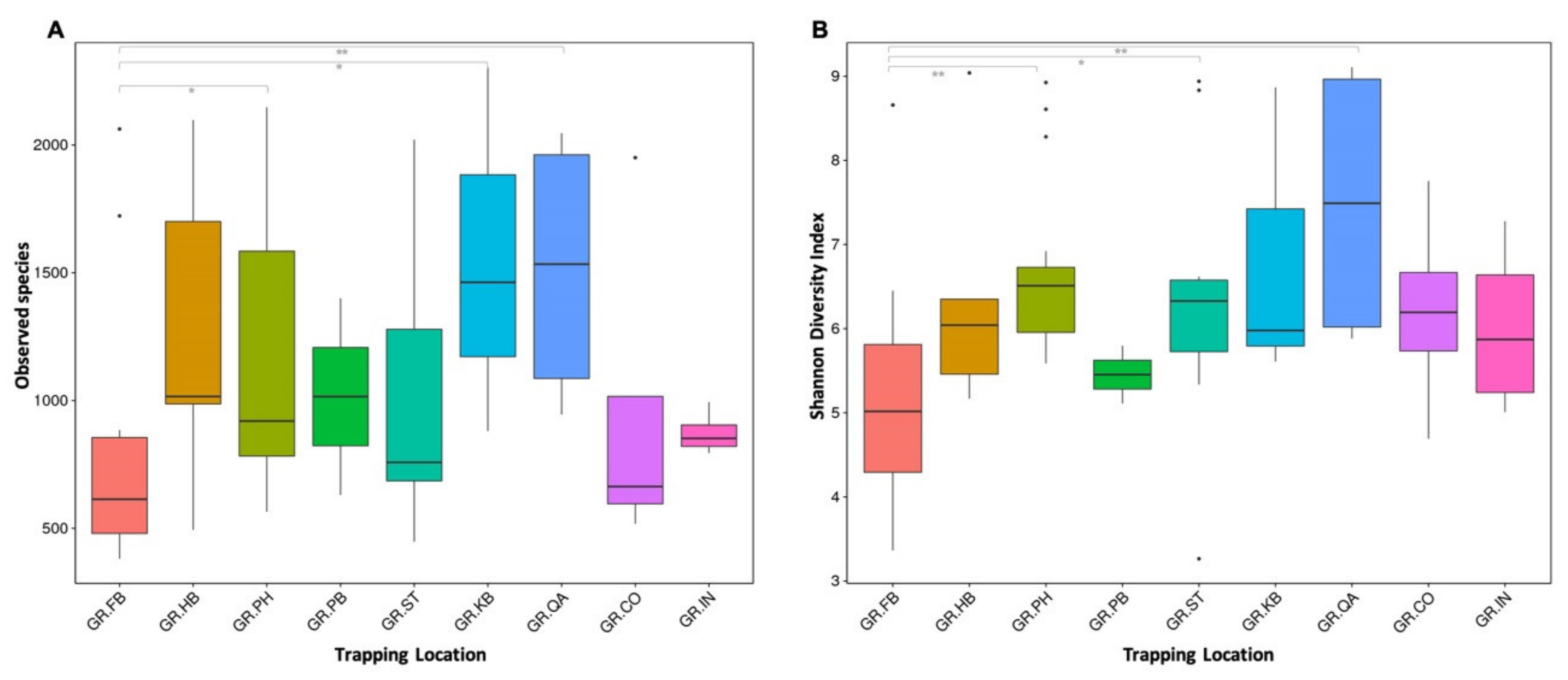
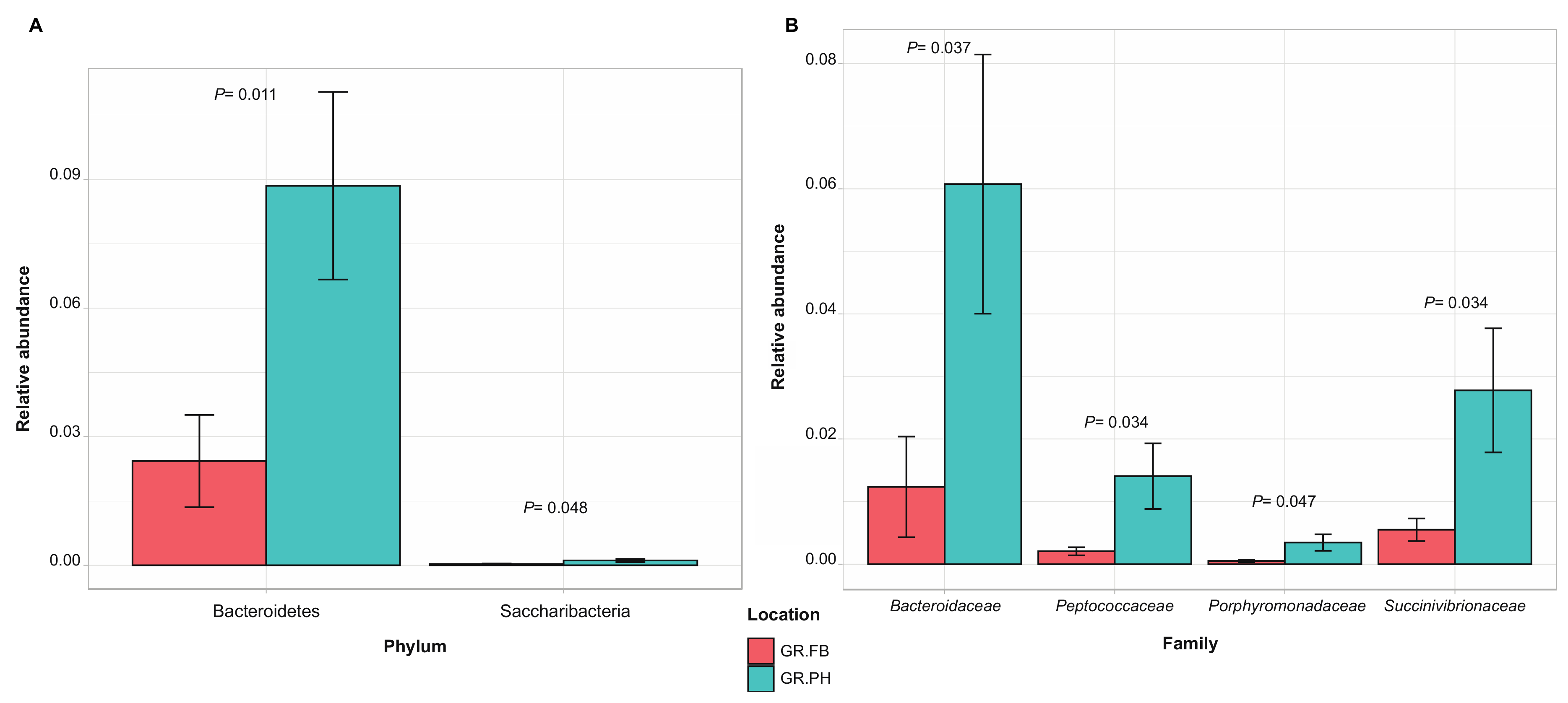
3.3. Biogeography and Sex-Related Patterns of Gut Microbial Diversity in Small Indian Mongooses
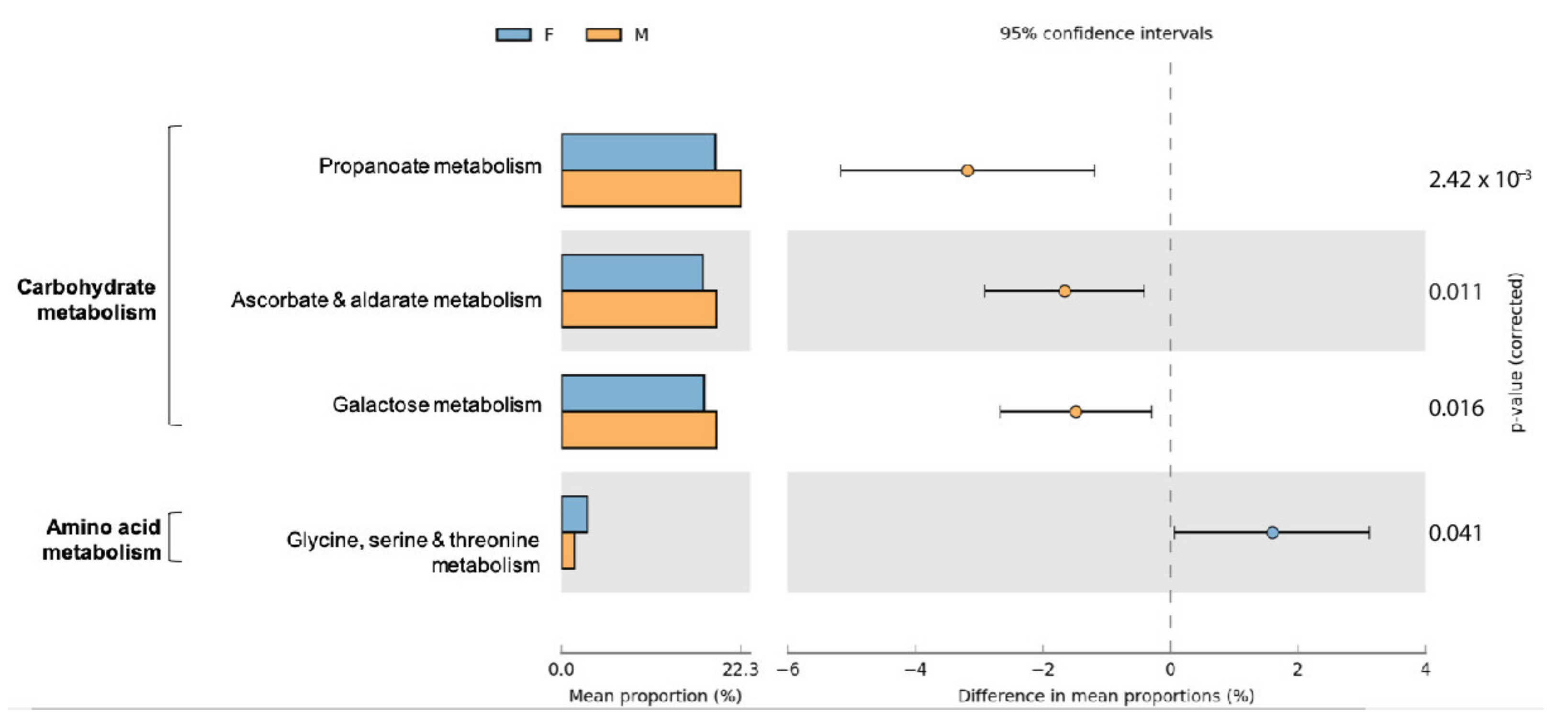
3.4. Functional Characterization of Bacterial Communities in Small Indian Mongooses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trevelline, B.K.; Fontaine, S.S.; Hartup, B.K.; Kohl, K.D. Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proc. R. Soc. B 2019, 286, 20182448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.G.; Waite, D.W.; Deines, P.; Bourne, D.G.; Digby, A.; Mckenzie, V.J.; Taylor, M.W. The microbiome in threatened species conservation. Biol. Conserv. 2019, 229, 85–98. [Google Scholar] [CrossRef]
- Global Invasive Species Database. 2020. Available online: http://www.iucngisd.org/gisd/100_worst.php (accessed on 29 April 2020).
- Veron, G.; Patou, M.L.; Pothet, G.; Simberloff, D.; Jennings, A.P. Systematic status and biogeography of the Javan and small Indian mongooses (Herpestidae, Carnivora). Zool. Scr. 2007, 36, 1–10. [Google Scholar] [CrossRef]
- Horst, G.R.; Hoagland, D.B.; Kilpatrick, C.W. The mongoose in the West Indies: The biogeography and population biology of an introduced species. In Biogeography of the West Indies: Patterns and Perspectives, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2001; pp. 409–424. [Google Scholar] [CrossRef]
- Hoagland, D.; Horst, G.; Kilpatrick, C. Biogeography and population biology of the mongoose in the West Indies. In Biogeography of the West Indies: Past, Present and Future; Wood, C., Ed.; Sandhill Crane Press: Gainesville, FL, USA, 1989; pp. 611–633. [Google Scholar]
- Cheng, T.; Halper, B.; Siebert, J.; Cruz-Martinez, L.; Chapwanya, A.; Kelly, P.; Ketzis, J.K.; Vessell, J.; Köster, L.; Yao, C. Parasites of small Indian mongoose, Herpestes auropunctatus, on St. Kitts, West Indies. Parasitol. Res. 2018, 117, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Berentsen, A.R.; Pitt, W.C.; Sugihara, R.T. Ecology of the small indian mongoose (herpestes auropunctatus) in North America. In Ecology and Management of Terrestrial Vertebrate Invasive Species in the United States; CRC Press: Boca Raton, FL, USA, 2017; pp. 251–268. [Google Scholar] [CrossRef]
- Louppe, V.; Leroy, B.; Herrel, A.; Veron, G. The globally invasive small Indian mongoose Urva auropunctata is likely to spread with climate change. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Shekhar, K. The status of mongooses in central India. Small Carniv. Conserv. 2003, 29, 22–23. [Google Scholar]
- Pieter, A.P.; Powers, K.E.; Hyzy, B.A. Initial explorations into the feeding ecology of the invasive small Indian mongoose in the Caribbean using stable isotope analyses. Bios 2016, 87, 155–162. [Google Scholar]
- Pereira, A.C.; Bandeira, V.; Fonseca, C.; Cunha, M.V. Egyptian mongoose (Herpestes ichneumon) gut microbiota: Taxonomical and functional differences across sex and age classes. Microorganisms 2020, 8, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patou, M.L.; Mclenachan, P.A.; Morley, C.G.; Couloux, A.; Jennings, A.P.; Veron, G. Molecular phylogeny of the Herpestidae (Mammalia, Carnivora) with a special emphasis on the Asian Herpestes. Mol. Phylogenet. Evol. 2009, 53, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Bandeira, V.; Virgos, E.; Carvalho, J.; Barros, T.; Cunha, M.V.; Fonseca, C. Diet footprint of Egyptian mongoose along ecological gradients: Effects of primary productivity and life history traits. Mamm. Biol. 2018, 88, 16–25. [Google Scholar] [CrossRef]
- Moeller, A.H.; Suzuki, T.A.; Lin, D.; Lacey, E.A.; Wasser, S.K.; Nachman, M.W. Dispersal limitation promotes the diversification of the mammalian gut microbiota. Proc. Natl. Acad. Sci. USA 2017, 114, 13768–13773. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [Green Version]
- Helmer, E.H.; Kennaway, T.A.; Pedreros, D.; Clark, M. Distributions of land cover and forest formations for St. Kitts, Nevis, St. Eustatius, Grenada and Barbados from satellite imagery. Caribb. J. Sci. 2008, 44, 175–198. [Google Scholar] [CrossRef]
- Kumar, J.; Kumar, M.; Gupta, S.; Ahmed, V.; Bhambi, M.; Pandey, R.; Chauhan, N.S. An Improved Methodology to Overcome Key Issues in Human Fecal Metagenomic DNA Extraction. Genom. Proteom. Bioinform. 2016, 14, 371–378. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.J.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high- throughput community sequencing data Intensity normalization improves color calling in SOLiD sequencing. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: http://www.r-project.org (accessed on 20 December 2020).
- Good, I.J. The population frequencies of species and the estimation of population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Youngblut, N.D.; Reischer, G.H.; Walters, W.; Schuster, N.; Walzer, C.; Stalder, G.; Ley, R.E.; Farnleitner, A.H. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, A.H.; Ochman, H. Rates of Gut Microbiome Divergence in Mammals. Mol. Ecol. 2018, 176, 139–148. [Google Scholar] [CrossRef]
- Vital, M.; Gao, J.; Rizzo, M.; Harrison, T.; Tiedje, J.M. Diet is a major factor governing the fecal butyrate-producing community structure across Mammalia, Aves and Reptilia. ISME J. 2015, 9, 832–843. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Fox, S.; Pemberton, D.; Hogg, C.; Papenfuss, A.T.; Belov, K. The Tasmanian devil microbiome-implications for conservation and management. Microbiome 2015, 3, 76. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, M.; Zhu, J.; Gao, Y.; Sha, W.; Ding, H.; Jiang, W.; Shenping, W. Age, gender, and feeding environment influence fecal microbial diversity in Spotted Hyenas (Crocuta crocuta). Curr. Microbiol. 2020. [Google Scholar] [CrossRef]
- Becker, A.A.M.J.; Hesta, M.; Hollants, J.; Janssens, G.P.J.; Huys, G. Phylogenetic analysis of faecal microbiota from captive cheetahs reveals underrepresentation of Bacteroidetes and Bifidobacteriaceae. BMC Microbiol. 2014, 14, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilla, R.; Suchodolski, J.S. The Role of the Canine Gut Microbiome and Metabolome in Health and Gastrointestinal Disease. Front. Vet. Sci. 2020, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanbari, M.; Kneifel, W.; Doming, K.J. A new view of the fish gut microbiome: Advances from next-generation sequencing. Aquaculture 2015, 448, 464–475. [Google Scholar] [CrossRef]
- Ferrario, C.; Statello, R.; Carnevali, L.; Mancabelli, L.; Milani, C.; Mangifesta, M.; Duranti, S.; Lugli, G.A.; Jimenez, B.; Lodge, S.; et al. How to Feed the Mammalian Gut Microbiota: Bacterial and Metabolic Modulation by Dietary Fibers. Front. Microbiol. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Kim, Y.S.; Unno, T.; Kim, B.Y.; Park, M.S. Sex differences in gut microbiota. World J. Mens. Health 2020, 38, 48–60. [Google Scholar] [CrossRef]
- Verbrugghe, A.; Hesta, M.; Daminet, S.; Polis, I.; Holst, J.J.; Buyse, J.; Wuyts, B.; Janssens, G.P.J. Propionate absorbed from the colon acts as gluconeogenic substrate in a strict carnivore, the domestic cat (Felis catus). J. Anim. Physiol. Anim. Nutr. 2012, 96, 1054–1064. [Google Scholar] [CrossRef]
- Kowalski, K.P.; Bacon, C.; Bickford, W.; Braun, H.; Clay, K.; Leduc-Lapierre, M.; Lillard, E.; McCormick, M.K.; Nelson, E.; Torres, M.; et al. Advancing the science of microbial symbiosis to support invasive species management: A case study on Phragmites in the Great Lakes. Front. Microbiol. 2015, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Shiokawa, K.; Llanes, A.; Hindoyan, A.; Cruz-Martinez, L.; Welcome, S.; Rajeev, S. Peridomestic small Indian mongoose: An invasive species posing as potential zoonotic risk for leptospirosis in the Caribbean. Acta Trop. 2019, 190, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Bahrndorff, S.; Alemu, T.; Alemneh, T.; Lund Nielsen, J. The Microbiome of Animals: Implications for Conservation Biology. Int. J. Genom. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, A.A.M.J.; Hill, K.; Butaye, P. Unraveling the Gut Microbiome of the Invasive Small Indian Mongoose (Urva auropunctata) in the Caribbean. Microorganisms 2021, 9, 465. https://doi.org/10.3390/microorganisms9030465
Becker AAMJ, Hill K, Butaye P. Unraveling the Gut Microbiome of the Invasive Small Indian Mongoose (Urva auropunctata) in the Caribbean. Microorganisms. 2021; 9(3):465. https://doi.org/10.3390/microorganisms9030465
Chicago/Turabian StyleBecker, Anne A. M. J., KC Hill, and Patrick Butaye. 2021. "Unraveling the Gut Microbiome of the Invasive Small Indian Mongoose (Urva auropunctata) in the Caribbean" Microorganisms 9, no. 3: 465. https://doi.org/10.3390/microorganisms9030465
APA StyleBecker, A. A. M. J., Hill, K., & Butaye, P. (2021). Unraveling the Gut Microbiome of the Invasive Small Indian Mongoose (Urva auropunctata) in the Caribbean. Microorganisms, 9(3), 465. https://doi.org/10.3390/microorganisms9030465