
The Nonribosomal Peptide Valinomycin: From Discovery to Bioactivity and Biosynthesis



Abstract
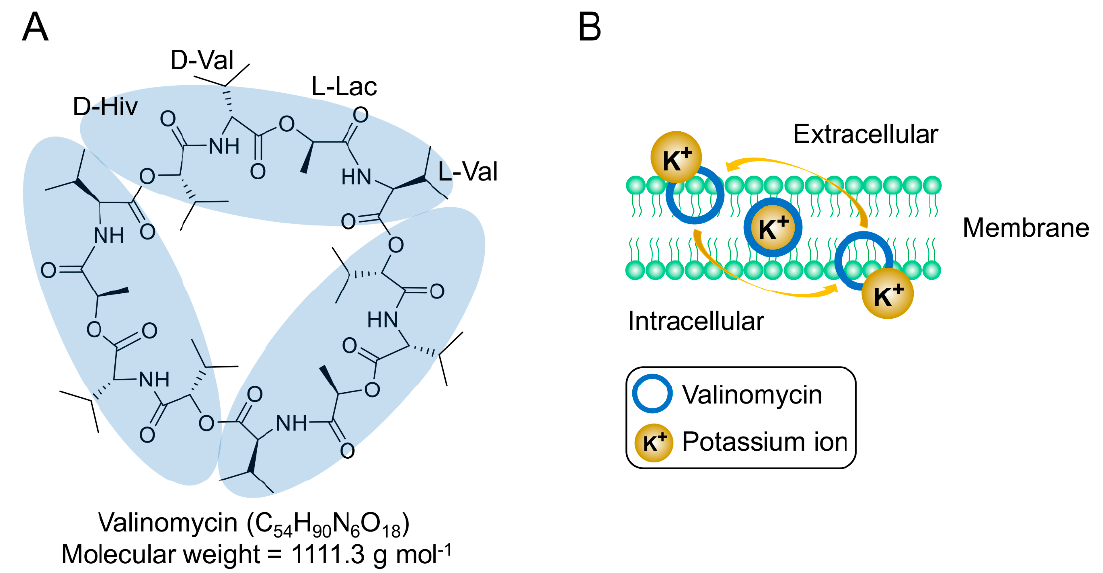
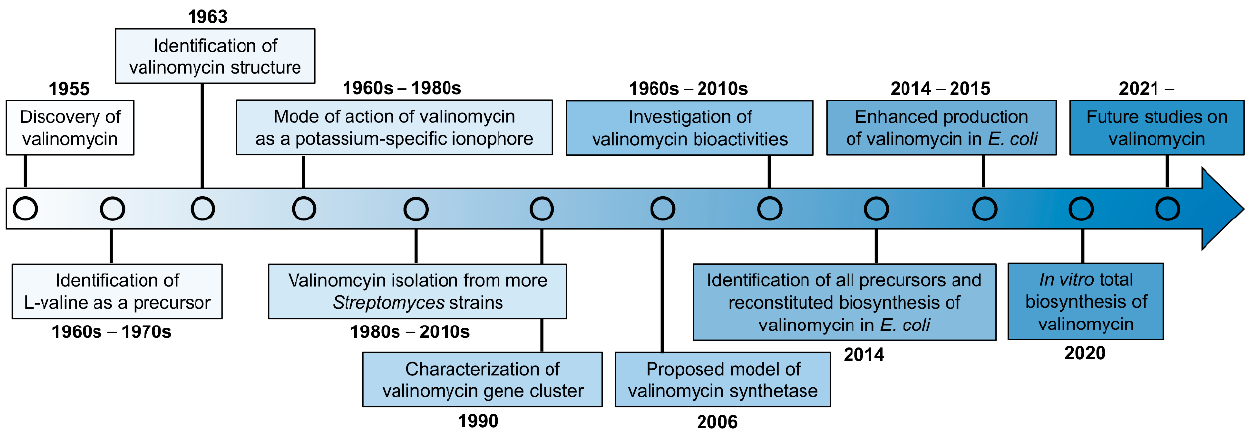
:1. Introduction
2. Biological Activities of Valinomycin
2.1. Antibacterial Activity
2.2. Antifungal Activity
2.3. Antiviral Activity
2.4. Insecticidal and Antiparasitic Activity
2.5. Antitumor Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioactivity | Efficacy a | Reference |
|---|---|---|
| Antibacterial | ||
| Streptococcus pyogenes | MIC 0.02 μg/mL | [44] |
| Clostridium sporogenes | MIC 8 μg/mL | [44] |
| Enterococcus faecalis | MIC 0.39–0.78 μg/disk | [42] |
| Streptococcus pneumoniae | MIC 0.39–0.78 μg/disk | [42] |
| Micrococcus luteus | MIC 25–50 μg/disk | [42] |
| Antifungal | ||
| Candida albicans | MIC 0.39–0.78 μg/disk | [42] |
| Cryptococcus neoformans | MIC 50–100 μg/disk | [42] |
| Phytophthora capsici | IC50 15.9 μg/mL | [47] |
| Botrytis cinerea | MIC 4 μg/mL | [48] |
| Magnaporthe grisea | MIC 4 μg/mL | [48] |
| Candida albicans | MIC 32 μg/mL | [48] |
| Colletotrichum gloeosporioides | MIC 256 μg/mL | [48] |
| Rhizoctonia solani | MIC 256 μg/mL | [48] |
| Penicillium verrucosum | IC50 0.005 ng/mL | [51] |
| Antiviral | ||
| Vesicular stomatitis virus (VSV) | GI90 10 μM | [42] |
| Severe acute respiratory syndrome coronavirus (SARS-CoV) | EC50 0.85 μM | [54] |
| Porcine reproductive and respiratory syndrome virus (PRRSV) | IC50 24 nM | [55] |
| Respiratory syncytial virus (RSV) | IC50 0.0015 μM | [63] |
| Middle East respiratory syndrome coronavirus (MERS-CoV) | IC50 84 nM | [56] |
| MERS-CoV | EC50 6.07 μM | [58] |
| MERS-CoV | IC50 5 nM | [57] |
| Human coronavirus OC43 (HCoV-OC43) | EC50 4.43 μM | [58] |
| Human coronavirus NL63 (HCoV-NL63) | EC50 1.89 μM | [58] |
| Mouse hepatitis virus A59 (MHV-A59) | EC50 6.78 μM | [58] |
| La Crosse virus (LACV) | IC50 588 nM | [57] |
| Rift Valley fever virus MP12 (RVFV MP-12) | IC50 41 nM | [57] |
| Human rhinovirus 2 (HRV2) | IC50 610 nM | [57] |
| Coxsackievirus B3 (CVB3) | IC50 971 nM | [57] |
| Zika virus (ZIKV) | IC50 78 nM | [57] |
| Keystone virus (KEYV) | IC50 156 nM | [57] |
| Human coronavirus 229E (HCoV-229E) | IC50 67 nM | [57] |
| Lassa virus (LASV) | EC50 0.61 μM | [62] |
| Lymphocytic choriomeningitis virus (LCMV) | EC50 0.15 μM | [62] |
| Insecticidal | ||
| Musca domestica (male) | LD50 0.02 μg | [70] |
| Musca domestica (female) | LD50 0.03 μg | [70] |
| Periplaneta americana (male) | LD50 0.19 μg | [70] |
| Periplaneta americana (female) | LD50 0.5 μg | [70] |
| Aedes aegypti | LC50 2–3 μg/mL | [65] |
| Tetranychus urticae | LC50 3 ppm | [65] |
| Epilachna varivestis | LC50 35 ppm | [65] |
| Plasmodium falciparum | IC50 5.3 ng/mL | [71] |
| Babesia gibsoni (in low potassium erythrocytes) | IC50 2.32 ng/mL | [72] |
| Babesia gibsoni (in high potassium erythrocytes) | IC50 570 ng/mL | [72] |
| Leishmania major | IC50 <0.11 μM | [68] |
| Trypanosoma brucei brucei | IC50 0.0032 μM | [68] |
| Antitumor | ||
| Human ovarian tumor cells CaOV-3 | IC50 0.1 nM | [74] |
| Murine P388 leukemia cancer cells | GI50 0.019 μg/mL | [42] |
| Human ovary OVCAR-3 tumor cells | GI50 1.9 × 10−4 μg/mL | [42] |
| Brain SF-295 tumor cells | GI50 3.5 × 10−4 μg/mL | [42] |
| Renal A-498 carcinoma cells | GI50 1.9 × 10−3 μg/mL | [42] |
| Lung NCI-H460 cancer cells | GI50 2.1 × 10−4 μg/mL | [42] |
| Colon KM20L2 carcinoma cells | GI50 2.7 × 10−4 μg/mL | [42] |
| Melanoma SK-MEL-5 cancer cells | GI50 2.6 × 10−4 μg/mL | [42] |
| Rat C6 glioma cells | IC50 0.0004 μM | [84] |
| Human A2780 ovarian carcinoma cells | IC50 2.18 μM | [84] |
| Human MCF-7 breast carcinoma cells | IC50 1.77 μM | [84] |
| Human HepG2 liver hepatocellular carcinoma cells | IC50 0.0008 μM | [84] |
| Human U251 glioma cells | IC50 7.6 nM | [85] |
3. Biogenesis of Valinomycin
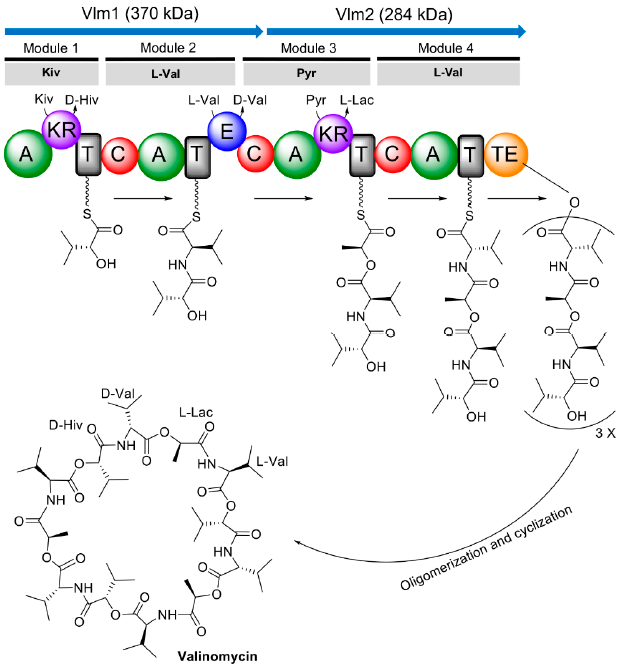
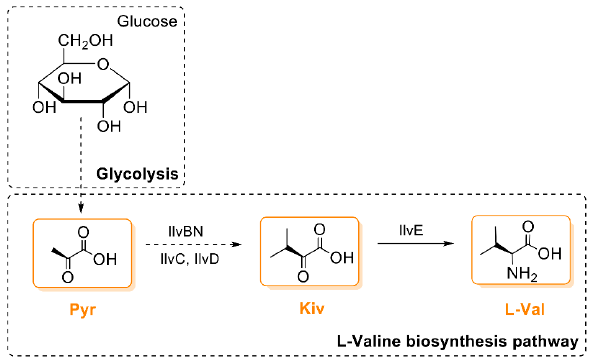
4. Biosynthesis of Valinomycin in Native Producers
5. Reconstituted Biosynthesis of Valinomycin In Vivo and In Vitro
5.1. Heterologous Production of Valinomycin in Escherichia coli
5.2. In Vitro Total Biosynthesis of Valinomycin
6. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Tang, Y. Natural Product Biosynthesis: Chemical Logic and Enzymatic Machinery; Royal Society of Chemistry Publishing: London, UK, 2017. [Google Scholar]
- Finking, R.; Marahiel, M.A. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 2004, 58, 453–488. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496. [Google Scholar] [CrossRef]
- Caboche, S.; Leclere, V.; Pupin, M.; Kucherov, G.; Jacques, P. Diversity of monomers in nonribosomal peptides: Towards the prediction of origin and biological activity. J. Bacteriol. 2010, 192, 5143–5150. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.T.; O’Brien, R.V.; Khosla, C. Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew. Chem. Int. Ed. 2013, 52, 7098–7124. [Google Scholar] [CrossRef] [Green Version]
- Felnagle, E.A.; Jackson, E.E.; Chan, Y.A.; Podevels, A.M.; Berti, A.D.; McMahon, M.D.; Thomas, M.G. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol. Pharm. 2008, 5, 191–211. [Google Scholar] [CrossRef]
- Marahiel, M.A. Working outside the protein-synthesis rules: Insights into non-ribosomal peptide synthesis. J. Pept. Sci. 2009, 15, 799–807. [Google Scholar] [CrossRef]
- Martín, J.F. New aspects of genes and enzymes for β-lactam antibiotic biosynthesis. Appl. Microbiol. Biotechnol. 1998, 50, 1–15. [Google Scholar] [CrossRef]
- Hubbard, B.K.; Walsh, C.T. Vancomycin assembly: Nature’s way. Angew. Chem. Int. Ed. 2003, 42, 730–765. [Google Scholar] [CrossRef]
- Brockmann, H.; Schmidt-Kastner, G. Valinomycin I, XXVII. Mitteil. über Antibiotica aus Actinomyceten. Chem. Ber. 1955, 88, 57–61. [Google Scholar] [CrossRef]
- Brockmann, H.; Geeren, H. Valinomycin II. Antibiotika aus Actinomyceten XXXVII. Die konstitution des Valinomycins. Justus Liebigs Ann. Chem. 1957, 603, 216–232. [Google Scholar] [CrossRef]
- Brockmann, H.; Springorum, M.; Träxler, G.; Höfer, I. Molekulargewicht des Valinomycins. Naturwissenschaften 1963, 50, 689. [Google Scholar] [CrossRef]
- Shemyakin, M.M.; Aldanova, N.A.; Vinogradova, E.I.; Feigina, M.Y. The structure and total synthesis of valinomycin. Tetrahedron Lett. 1963, 4, 1921–1925. [Google Scholar] [CrossRef]
- Sivanathan, S.; Scherkenbeck, J. Cyclodepsipeptides: A rich source of biologically active compounds for drug research. Molecules 2014, 19, 12368–12420. [Google Scholar] [CrossRef]
- Asher, I.M.; Rothschild, K.J.; Stanley, H.E. Raman spectroscopic study of the valinomycin-KSCN complex. J. Mol. Biol. 1974, 89, 205–222. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.A.; Sabesan, M.N.; Steinrauf, L.K. Crystal structure of valinomycin potassium picrate: Anion effects on valinomycin cation complexes. J. Am. Chem. Soc. 1981, 103, 5880–5885. [Google Scholar] [CrossRef]
- Neupert-Laves, K.; Dobler, M. The crystal structure of a K+ complex of valinomycin. Helv. Chim. Acta 1975, 58, 432–442. [Google Scholar] [CrossRef]
- Stillwell, W. Membrane transport. In An Introduction to Biological Membranes, 2nd ed.; Stillwell, W., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 440–442. [Google Scholar]
- Karle, I.L. Conformation of valinomycin in a triclinic crystal form. J. Am. Chem. Soc. 1975, 97, 4379–4386. [Google Scholar] [CrossRef]
- Karle, I.L.; Flippen-Anderson, J.L. A new conformation exhibiting near-threefold symmetry for uncomplexed valinomycin in crystals from dimethyl sulfoxide. J. Am. Chem. Soc. 1988, 110, 3253–3257. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. Polymorphic forms of valinomycin investigated by NMR crystallography. Int. J. Mol. Sci. 2020, 21, 4907. [Google Scholar] [CrossRef]
- Andreoli, T.E.; Tieffenberg, M.; Tosteson, D.C. The effect of valinomycin on the ionic permeability of thin lipid membranes. J. Gen. Physiol. 1967, 50, 2527–2545. [Google Scholar] [CrossRef]
- Junge, W.; Schmid, R. The mechanism of action of valinomycin on the thylakoid membrane: Characterization of the electric current density. J. Membr. Biol. 1971, 4, 179–192. [Google Scholar] [CrossRef]
- Pressman, B. Mechanism of action of transport-mediating antibiotics. Ann. N. Y. Acad. Sci. 1969, 147, 829–841. [Google Scholar] [CrossRef]
- Shemyakin, M.M.; Ovchinnikov, Y.A.; Ivanov, V.T.; Antonov, V.K.; Vinogradova, E.I.; Shkrob, A.M.; Malenkov, G.G.; Evstratov, A.V.; Laine, I.A.; Melnik, E.I.; et al. Cyclodepsipeptides as chemical tools for studying ionic transport through membranes. J. Membr. Biol. 1969, 1, 402–430. [Google Scholar] [CrossRef]
- Su, Z.F.; Ran, X.Q.; Leitch, J.J.; Schwan, A.L.; Faragher, R.; Lipkowski, J. How valinomycin ionophores enter and transport K+ across model lipid bilayer membranes. Langmuir 2019, 35, 16935–16943. [Google Scholar] [CrossRef]
- Tempelaars, M.H.; Rodrigues, S.; Abee, T. Comparative analysis of antimicrobial activities of valinomycin and cereulide, the Bacillus cereus emetic toxin. Appl. Environ. Microbiol. 2011, 77, 2755–2762. [Google Scholar] [CrossRef] [Green Version]
- Ovchinnikov, Y.A. Second FEBS-Ferdinand Springer lecture: Membrane active complexones. Chemistry and biological function. FEBS Lett. 1974, 44, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Shemyakin, M.M.; Vinogradova, E.I.; Feigina, M.Y.; Aldanova, N.A.; Loginova, N.F.; Ryabova, I.D.; Pavlenko, I.A. The structure-antimicrobial relation for valinomycin depsipeptides. Experientia 1965, 21, 548–552. [Google Scholar] [CrossRef]
- Shemyakin, M.M.; Vinogradova, E.I.; Ryabova, I.D.; Fonina, L.A.; Sanasaryan, A.A. Relationship between structure, stability of potassium complexes, and antimicrobial activity in a series of analogs of valinomycin. Chem. Nat. Compd. 1973, 9, 229–234. [Google Scholar] [CrossRef]
- Breitbart, H.; Herzberg, M. Membrane mediated inhibition of protein synthesis by valinomycin in reticulocytes. FEBS Lett. 1973, 32, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Breitbart, H.; Herzberg, M. Changes in energy charge and block of protein synthesis in rabbit reticulocytes under the action of valinomycin. Mol. Biol. Rep. 1980, 6, 195–198. [Google Scholar] [CrossRef]
- Breitbart, H.; Atlan, H.; Eltes, F.; Herzberg, M. Interaction between membrane properties and proteins synthesis in reticulocytes—A two step inhibition of protein synthesis by valinomycin. Mol. Biol. Rep. 1975, 2, 167–173. [Google Scholar] [CrossRef]
- Herzberg, M.; Breitbart, H. Block in the elongation of protein synthesis in rabbit reticulocyte by action of the ionophore valinomycin. Mol. Biol. Rep. 1980, 6, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Jakubkova, M.; Dzugasova, V.; Truban, D.; Abelovska, L.; Bhatia-Kissova, I.; Valachovic, M.; Klobucnikova, V.; Zeiselova, L.; Griac, P.; Nosek, J.; et al. Identification of yeast mutants exhibiting altered sensitivity to valinomycin and nigericin demonstrate pleiotropic effects of ionophores on cellular processes. PLoS ONE 2016, 11, e0164175. [Google Scholar] [CrossRef]
- Wieland, T. The History of Peptide Chemistry. In Peptide: Synthesis, Structure, and Applications; Gutte, B., Ed.; Academic Press: Cambridge, MA, USA, 1995; pp. 1–38. [Google Scholar]
- Rakovic, A.; Ziegler, J.; Mårtensson, C.U.; Prasuhn, J.; Shurkewitsch, K.; König, P.; Paulson, H.L.; Klein, C. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death. Differ. 2019, 26, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Li, S.; Han, T.L.; Zhou, F.; Zhang, X.; Tian, M.; Tang, L.; Li, Y. Study of mitophagy and ATP-related metabolomics based on β-amyloid levels in Alzheimer’s disease. Exp. Cell. Res. 2020, 396, 112266. [Google Scholar] [CrossRef] [PubMed]
- Harold, F.M.; Baarda, J.R. Gramicidin, valinomycin, and cation permeability of Streptococcus faecalis. J. Bacteriol. 1967, 94, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Pettit, G.R.; Tan, R.; Melody, N.; Kielty, J.M.; Pettit, R.K.; Herald, D.L.; Tucker, B.E.; Mallavia, L.P.; Doubek, D.L.; Schmidt, J.M. Antineoplastic agents. Part 409: Isolation and structure of montanastatin from a terrestrial actinomycete. Bioorg. Med. Chem. 1999, 7, 895–899. [Google Scholar] [CrossRef]
- Ryabova, I.D.; Gorneva, G.A.; Ovchinnikov, Y.A. Effect of valinomycin on ion transport in bacterial cells and on bacterial growth. Biochim. Biophys. Acta 1975, 401, 109–118. [Google Scholar] [CrossRef]
- Seshachalam, D.; Frahm, D.H.; Ferraro, F.M. Cation reversal of inhibition of growth by valinomycin in Streptococcus pyogenes and Clostridium sporogenes. Antimicrob. Agents Chemother. 1973, 3, 63–67. [Google Scholar] [CrossRef] [Green Version]
- Pagès, J.; James, C.; Winterhalter, M. The porin and the permeating antibiotic: A selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Pavlasova, E.; Harold, F.M. Energy coupling in the transport of beta-galactosides by Escherichia coli: Effect of proton conductors. J. Bacteriol. 1969, 98, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Lim, T.H.; Oh, H.C.; Kwon, S.Y.; Kim, J.H.; Seo, H.W.; Lee, J.H.; Kim, J.C.; Lim, C.H.; Cha, B.J.; Min, B.S. Antifungal activity of valinomycin, a cyclodepsipeptide from Streptomyces padanus TH-04. Nat. Prod. Sci. 2007, 13, 144–147. [Google Scholar]
- Park, C.N.; Lee, J.M.; Lee, D.; Kim, B.S. Antifungal activity of valinomycin, a peptide antibiotic produced by Streptomyces sp. strain M10 antagonistic to Botrytis cinerea. J. Microbiol. Biotechnol. 2008, 18, 880–884. [Google Scholar]
- Jeon, C.W.; Kim, D.R.; Kwak, Y.S. Valinomycin, produced by Streptomyces sp. S8, a key antifungal metabolite in large patch disease suppressiveness. World J. Microbiol. Biotechnol. 2019, 35, 128. [Google Scholar] [CrossRef]
- Ladeuze, S.; Lentz, N.; Delbrassinne, L.; Hu, X.; Mahillon, J. Antifungal activity displayed by cereulide, the emetic toxin produced by Bacillus cereus. Appl. Environ. Microbiol. 2011, 77, 2555–2558. [Google Scholar] [CrossRef] [Green Version]
- Mohd Danial, A.; Medina, A.; Sulyok, M.; Magan, N. Efficacy of metabolites of a Streptomyces strain (AS1) to control growth and mycotoxin production by Penicillium verrucosum, Fusarium verticillioides and Aspergillus fumigatus in culture. Mycotoxin Res. 2020, 36, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Makarasen, A.; Reukngam, N.; Khlaychan, P.; Chuysinuan, P.; Isobe, M.; Techasakul, S. Mode of action and synergistic effect of valinomycin and cereulide with amphotericin B against Candida albicans and Cryptococcus albidus. J. Mycol. Med. 2018, 28, 112–121. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, Z.; Chen, H.; Lu, Y.; Chen, X. Valinomycin as a potential antiviral agent against coronaviruses: A review. Biomed. J. 2020, 43, 414–423. [Google Scholar] [CrossRef]
- Wu, C.Y.; Jan, J.T.; Ma, S.H.; Kuo, C.J.; Juan, H.F.; Cheng, Y.S.; Hsu, H.H.; Huang, H.C.; Wu, D.; Brik, A.; et al. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karuppannan, A.K.; Wu, K.X.; Qiang, J.; Chu, J.J.; Kwang, J. Natural compounds inhibiting the replication of Porcine reproductive and respiratory syndrome virus. Antiviral Res. 2012, 94, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019, 10, 5770. [Google Scholar] [CrossRef] [PubMed]
- Sandler, Z.J.; Firpo, M.R.; Omoba, O.S.; Vu, M.N.; Menachery, V.D.; Mounce, B.C. Novel ionophores active against La Crosse virus identified through rapid antiviral screening. Antimicrob. Agents Chemother. 2020, 64, e00086-20. [Google Scholar] [CrossRef]
- Shen, L.; Niu, J.; Wang, C.; Huang, B.; Wang, W.; Zhu, N.; Deng, Y.; Wang, H.; Ye, F.; Cen, S.; et al. High-throughput screening and identification of potent broad-spectrum inhibitors of coronaviruses. J. Virol. 2019, 93, e00023-19. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Fatoki, T.H.; Ibraheem, O.; Ogunyemi, I.O.; Akinmoladun, A.C.; Ugboko, H.U.; Adeseko, C.J.; Awofisayo, O.A.; Olusegun, S.J.; Enibukun, J.M. Network analysis, sequence and structure dynamics of key proteins of coronavirus and human host, and molecular docking of selected phytochemicals of nine medicinal plants. J. Biomol. Struct. Dyn. 2020, 20, 1–23. [Google Scholar] [CrossRef]
- Cubitt, B.; Ortiz-Riano, E.; Cheng, B.Y.; Kim, Y.J.; Yeh, C.D.; Chen, C.Z.; Southall, N.O.E.; Zheng, W.; Martinez-Sobrido, L.; de la Torre, J.C. A cell-based, infectious-free, platform to identify inhibitors of lassa virus ribonucleoprotein (vRNP) activity. Antivir. Res. 2020, 173, 104667. [Google Scholar] [CrossRef]
- Norris, M.J.; Malhi, M.; Duan, W.; Ouyang, H.; Granados, A.; Cen, Y.; Tseng, Y.C.; Gubbay, J.; Maynes, J.; Moraes, T.J. Targeting intracellular ion homeostasis for the control of respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2018, 59, 733–744. [Google Scholar] [CrossRef]
- Wooding, M.; Naudé, Y.; Rohwer, E.; Bouwer, M. Controlling mosquitoes with semiochemicals: A review. Parasit Vectors 2020, 13, 80. [Google Scholar] [CrossRef]
- Heisey, R.M.; Huang, J.; Mishra, S.K.; Keller, J.E.; Miller, J.R.; Putnam, A.R.; D’Silva, T.D.J. Production of valinomycin, an insecticidal antibiotic, by Streptomyces griseus var. flexipertum var. nov. J. Agric. Food Chem. 1988, 36, 1283–1286. [Google Scholar] [CrossRef]
- Mishra, S.K.; Keller, J.E.; Miller, J.R.; Heisey, R.M.; Nair, M.G.; Putnam, A.R. Insecticidal and nematicidal properties of microbial metabolites. J. Ind. Microbiol. 1987, 2, 267–276. [Google Scholar] [CrossRef]
- Patterson, E.L.; Wright, D.P. Process for Controlling Insects, Nematodes and Mites Using Valinomycin. U.S. Patent No. 3,520,973, 21 July 1970. [Google Scholar]
- Pimentel-Elardo, S.M.; Kozytska, S.; Bugni, T.S.; Ireland, C.M.; Moll, H.; Hentschel, U. Anti-parasitic compounds from Streptomyces sp. strains isolated from Mediterranean sponges. Mar. Drugs 2010, 8, 373–380. [Google Scholar] [CrossRef]
- Angus, T.A. Similarity of effect of valinomycin and Bacillus thuringiensis parasporal protein in larvae of Bombyx mori. J. Invertebr. Pathol. 1968, 11, 145–146. [Google Scholar] [CrossRef]
- Pansa, M.C.; Natalizi, G.M.; Bettini, S. Toxicity of valinomycin on insects. J. Invertebr. Pathol. 1973, 22, 148–152. [Google Scholar] [CrossRef]
- Gumila, C.; Ancelin, M.L.; Jeminet, G.; Delort, A.M.; Miquel, G.; Vial, H.J. Differential in vitro activities of ionophore compounds against Plasmodium falciparum and mammalian cells. Antimicrob. Agents. Chemother. 1996, 40, 602–608. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, M.; Nakamura, K.; Tamura, N.; Hwang, S.J.; Yoshikawa, M.; Sasaki, N.; Ohta, H.; Yamato, O.; Maede, Y.; Takiguchi, M. Effects and mechanisms of action of ionophorous antibiotics valinomycin and salinomycin-Na on Babesia gibsoni in vitro. J. Parasitol. 2009, 95, 1532–1538. [Google Scholar] [CrossRef]
- Chen, D.; Song, M.; Mohamad, O.; Yu, S.P. Inhibition of Na+/K+-ATPase induces hybrid cell death and enhanced sensitivity to chemotherapy in human glioblastoma cells. BMC Cancer 2014, 14, 716. [Google Scholar] [CrossRef] [Green Version]
- Daoud, S.S.; Forde, N. Synergistic cytotoxic actions of cisplatin and liposomal valinomycin on human ovarian carcinoma cells. Cancer Chemother. Pharmacol. 1991, 28, 370–376. [Google Scholar] [CrossRef]
- Daoud, S.S.; Juliano, R.L. Reduced toxicity and enhanced antitumor effects in mice of the ionophoric drug valinomycin when incorporated in liposomes. Cancer Res. 1986, 46, 5518–5523. [Google Scholar] [PubMed]
- Inai, Y.; Yabuki, M.; Kanno, T.; Akiyama, J.; Yasuda, T.; Utsumi, K. Valinomycin induces apoptosis of ascites hepatoma cells (AH-130) in relation to mitochondrial membrane potential. Cell Struct. Funct. 1997, 22, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryoo, I.J.; Park, H.R.; Choo, S.J.; Hwang, J.H.; Park, Y.M.; Bae, K.H.; Shin-Ya, K.; Yoo, I.D. Selective cytotoxic activity of valinomycin against HT-29 human colon carcinoma cells via down-regulation of GRP78. Biol. Pharm. Bull. 2006, 29, 817–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.A.D.; Blaylock, M.G. Treatment of breast tumor cells in vitro with the mitochondrial membrane potential dissipater valinomycin increases 18F-FDG incorporation. J. Nucl. Med. 2007, 48, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Paananen, A.; Mikkola, R.; Sareneva, T.; Matikainen, S.; Andersson, M.; Julkunen, I.; Salkinoja-Salonen, M.S.; Timonen, T. Inhibition of human NK cell function by valinomycin, a toxin from Streptomyces griseus in indoor air. Infect. Immun. 2000, 68, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Paananen, A.; Järvinen, K.; Sareneva, T.; Salkinoja-Salonen, M.S.; Timonen, T.; Hölttä, E. Valinomycin-induced apoptosis of human NK cells is predominantly caspase independent. Toxicology 2005, 212, 37–45. [Google Scholar] [CrossRef]
- Abdalah, R.; Wei, L.; Francis, K.; Yu, S.P. Valinomycin-induced apoptosis in Chinese hamster ovary cells. Neurosci. Lett. 2006, 405, 68–73. [Google Scholar] [CrossRef]
- Deckers, C.L.P.; Lyons, A.B.; Samuel, K.; Sanderson, A.; Maddy, A.H. Alternative pathways of apoptosis induced by methylprednisolone and valinomycin analyzed by flow cytometry. Exp. Cell Res. 1993, 208, 362–370. [Google Scholar] [CrossRef]
- Furlong, I.J.; Lopez Mediavilla, C.; Ascaso, R.; Lopez Rivas, A.; Collins, M.K. Induction of apoptosis by valinomycin: Mitochondrial permeability transition causes intracellular acidification. Cell Death. Differ. 1998, 5, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Iacobazzi, R.M.; Annese, C.; Azzariti, A.; D’Accolti, L.; Franco, M.; Fusco, C.; La Piana, G.; Laquintana, V.; Denora, N. Antitumor potential of conjugable valinomycins bearing hydroxyl sites: In vitro studies. ACS Med. Chem. Lett. 2013, 4, 1189–1192. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Anjum, K.; Song, T.; Wang, W.; Liang, Y.; Chen, M.; Huang, H.; Lian, X.Y.; Zhang, Z. Antiproliferative cyclodepsipeptides from the marine actinomycete Streptomyces sp. P11-23B downregulating the tumor metabolic enzymes of glycolysis, glutaminolysis, and lipogenesis. Phytochemistry 2017, 135, 151–159. [Google Scholar] [CrossRef]
- MacDonald, J.C. Biosynthesis of valinomycin. Can. J. Microbiol. 1960, 6, 27–34. [Google Scholar] [CrossRef]
- MacDonald, J.C.; Slater, G.P. Biosynthesis of valinomycin. Can. J. Biochem. 1968, 46, 573–578. [Google Scholar] [CrossRef]
- Ristow, H.; Salnikow, J.; Kleinkauf, H. Biosynthesis of valinomycin. FEBS Lett. 1974, 42, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Anke, T.; Lipmann, F. Studies on the biosynthesis of valinomycin. FEBS Lett. 1977, 82, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Perkins, J.B.; Guterman, S.K.; Howitt, C.L.; Williams, V.E.; Pero, J. Streptomyces genes involved in biosynthesis of the peptide antibiotic valinomycin. J. Bacteriol. 1990, 172, 3108–3116. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.Q. Deciphering the biosynthetic codes for the potent anti-SARS-CoV cyclodepsipeptide valinomycin in Streptomyces tsusimaensis ATCC 15141. ChemBioChem 2006, 7, 471–477. [Google Scholar] [CrossRef]
- Magarvey, N.A.; Ehling-Schulz, M.; Walsh, C.T. Characterization of the cereulide NRPS α-hydroxy acid specifying modules: Activation of α-keto acids and chiral reduction on the assembly line. J. Am. Chem. Soc. 2006, 128, 10698–10699. [Google Scholar] [CrossRef]
- Jaitzig, J.; Li, J.; Süssmuth, R.D.; Neubauer, P. Reconstituted biosynthesis of the nonribosomal macrolactone antibiotic valinomycin in Escherichia coli. ACS Synth. Biol. 2014, 3, 432–438. [Google Scholar] [CrossRef]
- Huguenin-Dezot, N.; Alonzo, D.A.; Heberlig, G.W.; Mahesh, M.; Nguyen, D.P.; Dornan, M.H.; Boddy, C.N.; Schmeing, T.M.; Chin, J.W. Trapping biosynthetic acyl-enzyme intermediates with encoded 2,3-diaminopropionic acid. Nature 2019, 565, 112–117. [Google Scholar] [CrossRef]
- Andersson, M.A.; Mikkola, R.; Kroppenstedt, R.M.; Rainey, F.A.; Salkinoja-Salonen, M.S. The mitochondrial toxin produced by Streptomyces griseus strains isolated from an indoor environment is valinomycin. Appl. Environ. Microbiol. 1998, 64, 4767–4773. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Jamwal, V.; Singh, V.P.; Wazir, P.; Awasthi, P.; Singh, D.; Vishwakarma, R.A.; Gandhi, S.G.; Chaubey, A. Revelation and cloning of valinomycin synthetase genes in Streptomyces lavendulae ACR-DA1 and their expression analysis under different fermentation and elicitation conditions. J. Biotechnol. 2017, 253, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Wulff, E.G.; Mguni, C.M.; Mansfeld-Giese, K.; Fels, J.; Lübeck, M.; Hockenhull, J. Biochemical and molecular characterization of Bacillus amyloliquefaciens, B. subtilis and B. pumilus isolates with distinct antagonistic potential against Xanthomonas campestris pv. campestris. Plant Pathol. 2002, 51, 574–584. [Google Scholar] [CrossRef]
- Gaiser, R.A.; Medema, M.H.; Kleerebezem, M.; van Baarlen, P.; Wells, J.M. Draft Genome sequence of a porcine commensal, Rothia nasimurium, encoding a nonribosomal peptide synthetase predicted to produce the ionophore antibiotic valinomycin. Genome Announc. 2017, 5, e00453-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matter, A.M.; Hoot, S.B.; Anderson, P.D.; Neves, S.S.; Cheng, Y.Q. Valinomycin biosynthetic gene cluster in Streptomyces: Conservation, ecology and evolution. PLoS ONE 2009, 4, e7194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, P.R. Natural products and the gene cluster revolution. Trends Microbiol. 2016, 24, 968–977. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Ng, B.G.; Kim, B.S. Increased valinomycin production in mutants of Streptomyces sp. M10 defective in bafilomycin biosynthesis and branched-chain α-keto acid dehydrogenase complex expression. J. Ind. Microbiol. Biotechnol. 2015, 42, 1507–1517. [Google Scholar] [CrossRef]
- Singh, V.P.; Sharma, R.; Sharma, V.; Raina, C.; Kapoor, K.K.; Kumar, A.; Chaubey, A.; Singh, D.; Vishwakarma, R.A. Isolation of depsipeptides and optimization for enhanced production of valinomycin from the North-Western Himalayan cold desert strain Streptomyces lavendulae. J. Antibiot. 2019, 72, 617–624. [Google Scholar] [CrossRef]
- Myronovskyi, M.; Luzhetskyy, A. Heterologous production of small molecules in the optimized Streptomyces hosts. Nat. Prod. Rep. 2019, 36, 1281–1294. [Google Scholar] [CrossRef]
- Nepal, K.K.; Wang, G. Streptomycetes: Surrogate hosts for the genetic manipulation of biosynthetic gene clusters and production of natural products. Biotechnol. Adv. 2019, 37, 1–20. [Google Scholar] [CrossRef]
- Li, J.; Neubauer, P. Escherichia coli as a cell factory for heterologous production of nonribosomal peptides and polyketides. N. Biotechnol. 2014, 31, 579–585. [Google Scholar] [CrossRef]
- Luo, Y.; Li, B.Z.; Liu, D.; Zhang, L.; Chen, Y.; Jia, B.; Zeng, B.X.; Zhao, H.; Yuan, Y.J. Engineered biosynthesis of natural products in heterologous hosts. Chem. Soc. Rev. 2015, 44, 5265–5290. [Google Scholar] [CrossRef]
- Zhang, H.; Boghigian, B.A.; Armando, J.; Pfeifer, B.A. Methods and options for the heterologous production of complex natural products. Nat. Prod. Rep. 2011, 28, 125–151. [Google Scholar] [CrossRef]
- Huo, L.; Hug, J.J.; Fu, C.; Bian, X.; Zhang, Y.; Müller, R. Heterologous expression of bacterial natural product biosynthetic pathways. Nat. Prod. Rep. 2019, 36, 1412–1436. [Google Scholar] [CrossRef]
- Lambalot, R.H.; Gehring, A.M.; Flugel, R.S.; Zuber, P.; LaCelle, M.; Marahiel, M.A.; Reid, R.; Khosla, C.; Walsh, C.T. A new enzyme superfamily—the phosphopantetheinyl transferases. Chem. Biol. 1996, 3, 923–936. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jaitzig, J.; Hillig, F.; Süssmuth, R.; Neubauer, P. Enhanced production of the nonribosomal peptide antibiotic valinomycin in Escherichia coli through small-scale high cell density fed-batch cultivation. Appl. Microbiol. Biotechnol. 2014, 98, 591–601. [Google Scholar] [CrossRef]
- Krause, M.; Ukkonen, K.; Haataja, T.; Ruottinen, M.; Glumoff, T.; Neubauer, A.; Neubauer, P.; Vasala, A. A novel fed-batch based cultivation method provides high cell-density and improves yield of soluble recombinant proteins in shaken cultures. Microb. Cell Fact. 2010, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Panula-Perälä, J.; Siurkus, J.; Vasala, A.; Wilmanowski, R.; Casteleijn, M.G.; Neubauer, P. Enzyme controlled glucose auto-delivery for high cell density cultivations in microplates and shake flasks. Microb. Cell Fact. 2008, 7, 31. [Google Scholar] [CrossRef] [Green Version]
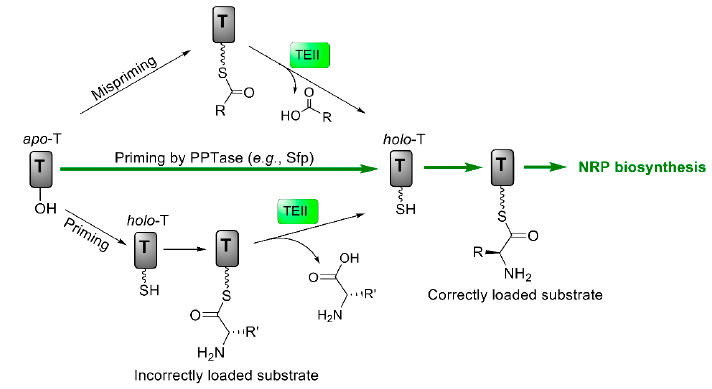
- Kotowska, M.; Pawlik, K. Roles of type II thioesterases and their application for secondary metabolite yield improvement. Appl. Microbiol. Biotechnol. 2014, 98, 7735–7746. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, D.; Mootz, H.D.; Linne, U.; Marahiel, M.A. Regeneration of misprimed nonribosomal peptide synthetases by type II thioesterases. Proc. Natl. Acad. Sci. USA 2002, 99, 14083–14088. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.; Kohli, R.M.; Bruner, S.D.; Walsh, C.T. Type II thioesterase restores activity of a NRPS module stalled with an aminoacyl-S-enzyme that cannot be elongated. ChemBioChem 2004, 5, 1290–1293. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaitzig, J.; Theuer, L.; Legala, O.E.; Süssmuth, R.D.; Neubauer, P. Type II thioesterase improves heterologous biosynthesis of valinomycin in Escherichia coli. J. Biotechnol. 2015, 193, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaitzig, J.; Lu, P.; Süssmuth, R.D.; Neubauer, P. Scale-up bioprocess development for production of the antibiotic valinomycin in Escherichia coli based on consistent fed-batch cultivations. Microb. Cell Fact. 2015, 14, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bundy, B.C.; Hunt, J.P.; Jewett, M.C.; Swartz, J.R.; Wood, D.W.; Frey, D.D.; Rao, G. Cell-free biomanufacturing. Curr. Opin. Chem. Eng. 2018, 22, 177–183. [Google Scholar] [CrossRef]
- Li, J.; Zhang, L.; Liu, W. Cell-free synthetic biology for in vitro biosynthesis of pharmaceutical natural products. Synth. Syst. Biotechnol. 2018, 3, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Q.; Zhang, L.; Chen, M.; Li, J. Cell-free protein synthesis: Recent advances in bacterial extract sources and expanded applications. Biochem. Eng. J. 2019, 141, 182–189. [Google Scholar] [CrossRef]
- Silverman, A.D.; Karim, A.S.; Jewett, M.C. Cell-free gene expression systems: An expanding repertoire of applications. Nat. Rev. Genet. 2020, 21, 151–170. [Google Scholar] [CrossRef]
- Swartz, J.R. Expanding biological applications using cell-free metabolic engineering: An overview. Metab. Eng. 2018, 50, 156–172. [Google Scholar] [CrossRef]
- Dudley, Q.M.; Karim, A.S.; Nash, C.J.; Jewett, M.C. In vitro prototyping of limonene biosynthesis using cell-free protein synthesis. Metab. Eng. 2020, 61, 251–260. [Google Scholar] [CrossRef]
- Goering, A.W.; Li, J.; McClure, R.A.; Thomson, R.J.; Jewett, M.C.; Kelleher, N.L. In vitro reconstruction of nonribosomal peptide biosynthesis directly from DNA using cell-free protein synthesis. ACS Synth. Biol. 2017, 6, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Grubbe, W.S.; Rasor, B.J.; Krüger, A.; Jewett, M.C.; Karim, A.S. Cell-free styrene biosynthesis at high titers. Metab. Eng. 2020, 61, 89–95. [Google Scholar] [CrossRef]
- Karim, A.S.; Jewett, M.C. A cell-free framework for rapid biosynthetic pathway prototyping and enzyme discovery. Metab. Eng. 2016, 36, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Karim, A.S.; Dudley, Q.M.; Juminga, A.; Yuan, Y.; Crowe, S.A.; Heggestad, J.T.; Garg, S.; Abdalla, T.; Grubbe, W.S.; Rasor, B.J.; et al. In vitro prototyping and rapid optimization of biosynthetic enzymes for cellular design. Nat. Chem. Biol. 2020, 16, 912–919. [Google Scholar] [CrossRef]
- Feng, J.; Yang, C.; Zhao, Z.; Xu, J.; Li, J.; Li, P. Application of cell-free protein synthesis system for the biosynthesis of L-theanine. ACS Synth. Biol. 2021, 10, 620–631. [Google Scholar] [CrossRef]
- Liu, W.Q.; Wu, C.; Jewett, M.C.; Li, J. Cell-free protein synthesis enables one-pot cascade biotransformation in an aqueous-organic biphasic system. Biotechnol. Bioeng. 2020, 117, 4001–4008. [Google Scholar] [CrossRef]
- Zhuang, L.; Huang, S.; Liu, W.Q.; Karim, A.S.; Jewett, M.C.; Li, J. Total in vitro biosynthesis of the nonribosomal macrolactone peptide valinomycin. Metab. Eng. 2020, 60, 37–44. [Google Scholar] [CrossRef]
- Li, J.; Lawton, T.J.; Kostecki, J.S.; Nisthal, A.; Fang, J.; Mayo, S.L.; Rosenzweig, A.C.; Jewett, M.C. Cell-free protein synthesis enables high yielding synthesis of an active multicopper oxidase. Biotechnol. J. 2016, 11, 212–218. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, H.; Kwon, Y.C.; Jewett, M.C. Establishing a high yielding Streptomyces-based cell-free protein synthesis system. Biotechnol. Bioeng. 2017, 114, 1343–1353. [Google Scholar] [CrossRef]
- Li, J.; Wang, H.; Jewett, M.C. Expanding the palette of Streptomyces-based cell-free protein synthesis systems with enhanced yields. Biochem. Eng. J. 2018, 130, 29–33. [Google Scholar] [CrossRef]
- Moore, S.J.; Lai, H.E.; Needham, H.; Polizzi, K.M.; Freemont, P.S. Streptomyces venezuelae TX-TL—A next generation cell-free synthetic biology tool. Biotechnol. J. 2017, 12, 1600678. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Liu, W.Q.; Li, J. Translation related factors improve the productivity of a Streptomyces-based cell-free protein synthesis system. ACS Synth. Biol. 2020, 9, 1221–1224. [Google Scholar] [CrossRef]
- Zawada, J.F.; Yin, G.; Steiner, A.R.; Yang, J.; Naresh, A.; Roy, S.M.; Gold, D.S.; Heinsohn, H.G.; Murray, C.J. Microscale to manufacturing scale-up of cell-free cytokine production—A new approach for shortening protein production development timelines. Biotechnol. Bioeng. 2011, 108, 1570–1578. [Google Scholar] [CrossRef] [Green Version]





| Production | Yield a | Note | Reference |
|---|---|---|---|
| Native host | |||
| Streptomyces fulvissimus | 17 mg/L | Static cultivation in flask, 20 d | [12] |
| Streptomyces sp. PRL1642 | 50–58 mg/L | Shake-flask, 3–5 d | [86] |
| Streptomyces griseus var. flexipertum | N.R. | Bioreactor, 5 d | [65] |
| Streptomyces levoris A-9 | N.R. | Shake-flask, 5 d | [90] |
| Streptomyces griseus strains 2/ppi, 8/ppi, 10/ppi, and 1/k | 0.6–1.4 mg/mg wet biomass | Cultivation on agar plates, 10–12 d | [95] |
| Streptomyces anulatus (Montana) | 4.65 mg/L | Shake-flask, 3 d | [42] |
| Streptomyces anulatus (Malaysian) | 1.5 mg/L | Shake-flask, 3 d | [42] |
| Streptomyces exfoliatus (Malaysian) | 3.9 mg/L | Shake-flask, 3 d | [42] |
| Streptomyces padanus TH-04 | 70 mg/L | Shake-flask, 7 d | [47] |
| Streptomyces sp. M10 | 3. 83 mg/L | Shake-flask, 2 d | [48] |
| Streptomyces sp. M10 | 16 mg/L | Shake-flask, 3 d, engineered M10 strain | [101] |
| Streptomyces tsusimaensis | 8.45 mg/L | Shake-flask, 6 d | [99] |
| Streptomyces griseus 1/k | 10.19 mg/L | Shake-flask, 6 d | [99] |
| Streptomyces griseus 10/ppi | 22.08 mg/L | Shake-flask, 6 d | [99] |
| Streptomyces sp. 22 | N.R. | Cultivation on agar plates, 7 d | [68] |
| Streptomyces sp. 34 | N.R. | Cultivation on agar plates, 7 d | [68] |
| Streptomyces sp. P11–23B | 0.74 mg/L | Shake-flask, 14 d | [85] |
| Streptomyces lavendulae ACR-DA1 | 84 mg/L | Shake-flask, 8 d, l-valine feeding | [96] |
| Streptomyces lavendulae ACR-DA1 | 19.4 mg/L | Bioreactor, 8 d | [102] |
| Streptomyces sp. S8 | N.R. | Shake-flask, 14 d | [49] |
| Streptomyces parvus | 0.15 mg/g dry biomass | Shake-flask, 5 d | [51] |
| Bacillus subtilis | N.R. | Shake-flask, 3 d | [97] |
| Bacillus pumilus | N.R. | Shake-flask, 3 d | [97] |
| Bacillus amyloliquefaciens | N.R. | Shake-flask, 3 d | [97] |
| Rothia nasimurium | N.R. | Genome sequencing and prediction | [98] |
| Heterologous host | |||
| Escherichia coli | 0.3 mg/L | Shake-flask, 36 h | [93] |
| Escherichia coli | 6.4 mg/L | 24-well plate, 48 h | [110] |
| Escherichia coli | 10 mg/L | Shake-flask, fed-batch cultivation, 48 h | [110] |
| Escherichia coli | 2 mg/L | Bioreactor, 48 h | [117] |
| Escherichia coli | 13 mg/L | Shake-flask, 48 h, coexpression of TEII | [116] |
| In vitro system | |||
| Escherichia coli cell-free system | 37 μg/L | CFPS, 20 h | [130] |
| Escherichia coli cell-free system | 77 μg/L | CFME, 12 h | [130] |
| Escherichia coli cell-free system | 30 mg/L | CFPS-ME, 15 h | [130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Liu, Y.; Liu, W.-Q.; Neubauer, P.; Li, J. The Nonribosomal Peptide Valinomycin: From Discovery to Bioactivity and Biosynthesis. Microorganisms 2021, 9, 780. https://doi.org/10.3390/microorganisms9040780
Huang S, Liu Y, Liu W-Q, Neubauer P, Li J. The Nonribosomal Peptide Valinomycin: From Discovery to Bioactivity and Biosynthesis. Microorganisms. 2021; 9(4):780. https://doi.org/10.3390/microorganisms9040780
Chicago/Turabian StyleHuang, Shuhui, Yushi Liu, Wan-Qiu Liu, Peter Neubauer, and Jian Li. 2021. "The Nonribosomal Peptide Valinomycin: From Discovery to Bioactivity and Biosynthesis" Microorganisms 9, no. 4: 780. https://doi.org/10.3390/microorganisms9040780
APA StyleHuang, S., Liu, Y., Liu, W.-Q., Neubauer, P., & Li, J. (2021). The Nonribosomal Peptide Valinomycin: From Discovery to Bioactivity and Biosynthesis. Microorganisms, 9(4), 780. https://doi.org/10.3390/microorganisms9040780