
Deciphering the Endophytic and Rhizospheric Microbial Communities of a Metallophyte Commelina communis in Different Cu-Polluted Soils

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant and Soil Sample Collection
2.2. Soil Properties Analysis
2.3. Endosphere and Rhizosphere Sample Preparation
2.4. DNA Extraction and 16S rRNA Sequencing
2.5. Bioinformatic Analyses
2.6. Statistical Analyses
3. Results
3.1. Physico-Chemical Properties of Rhizosphere Soils
3.2. General Features of the Sequencing Data
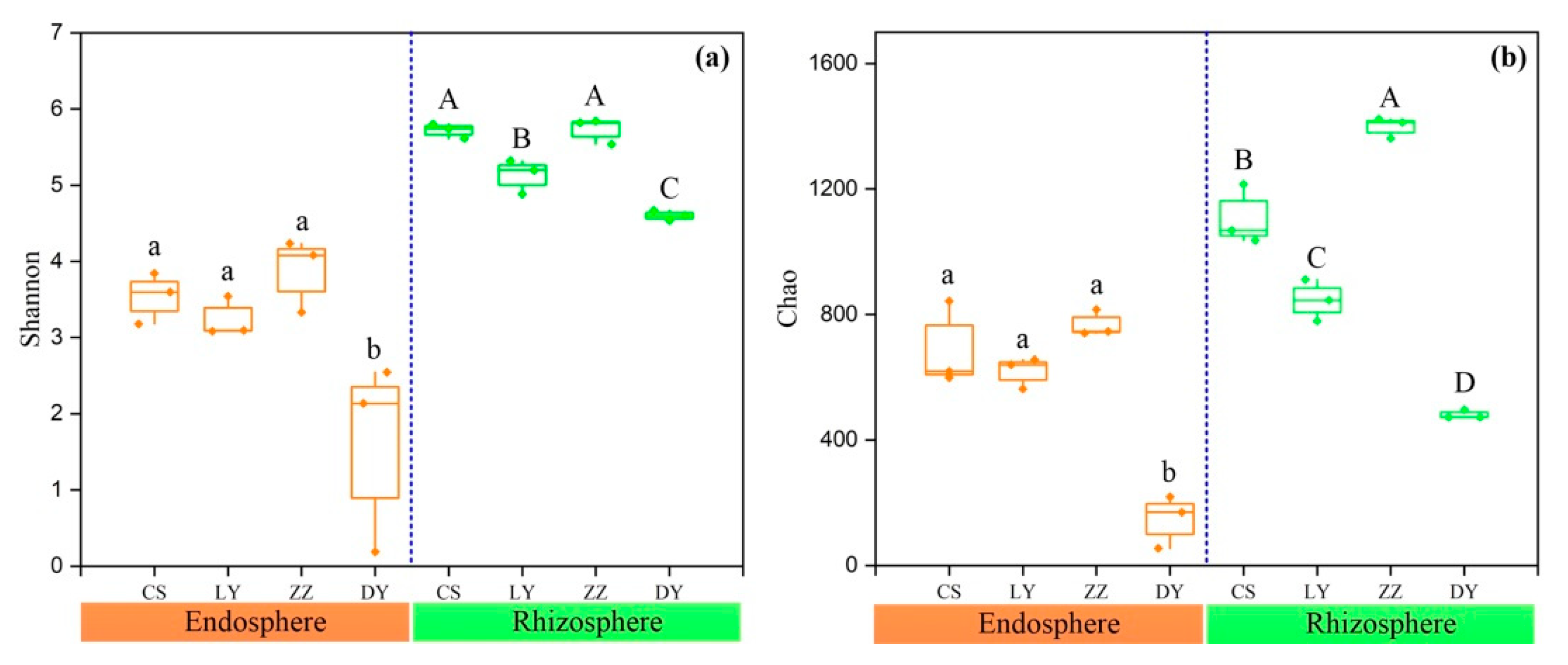
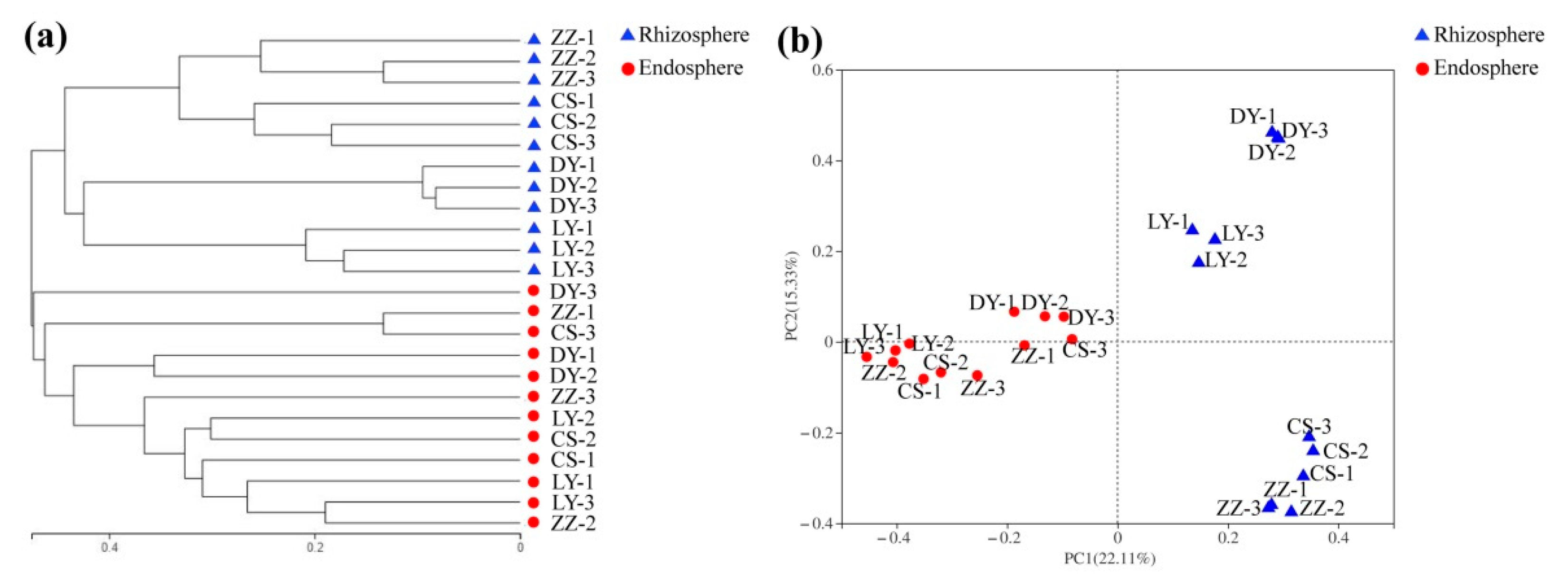
3.3. Diversity and Structure of the Endosphere and Rhizosphere Bacteria Community
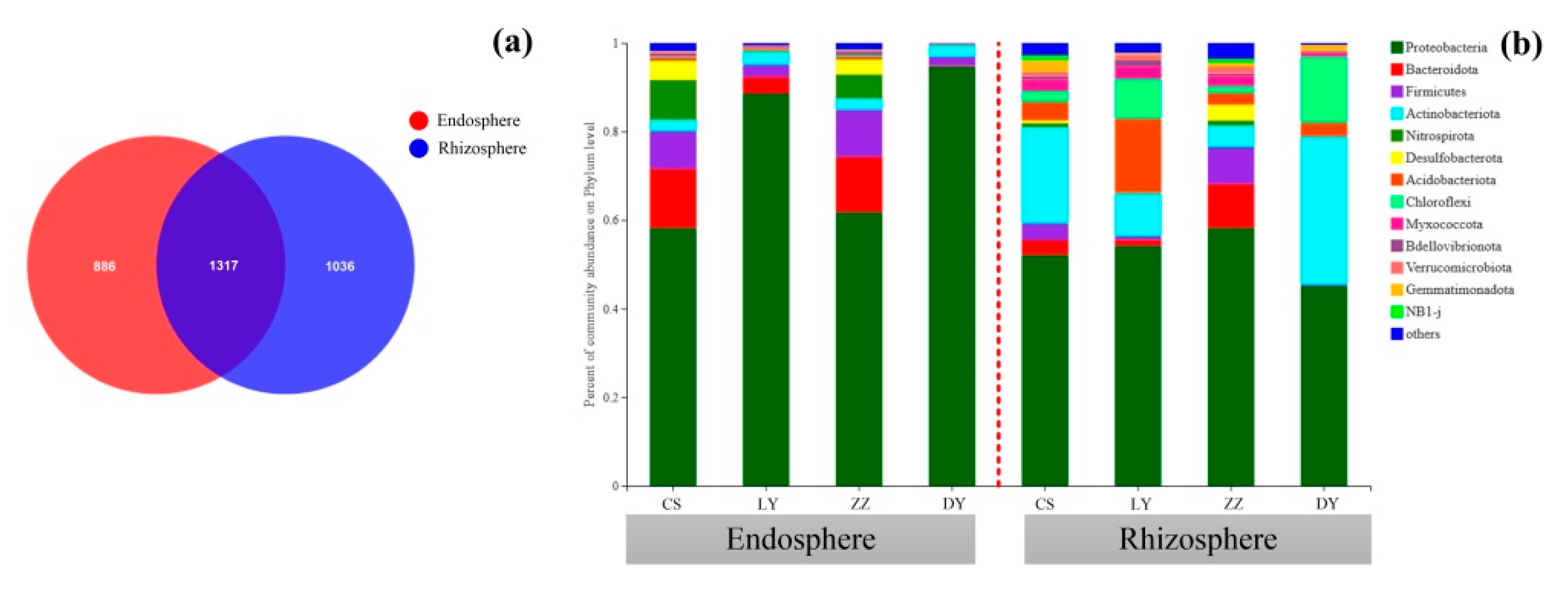
3.4. Venn Diagram for Rhizospheric and Endophytic Bacterial Communities
3.5. Taxonomic Distributions of the Endosphere and Rhizosphere Bacteria Community
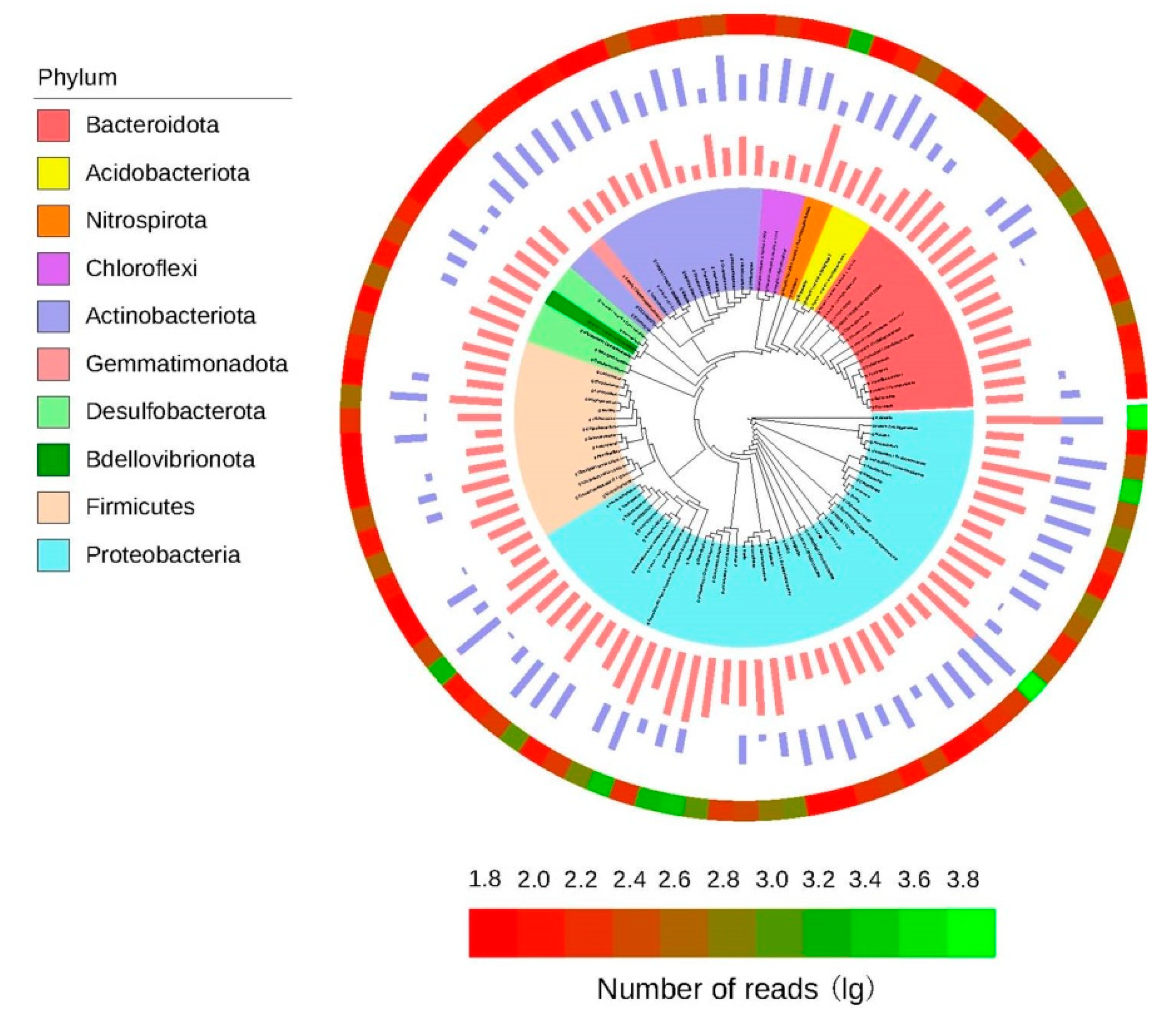
3.6. Phylogenetic Tree of the Core Microorganisms
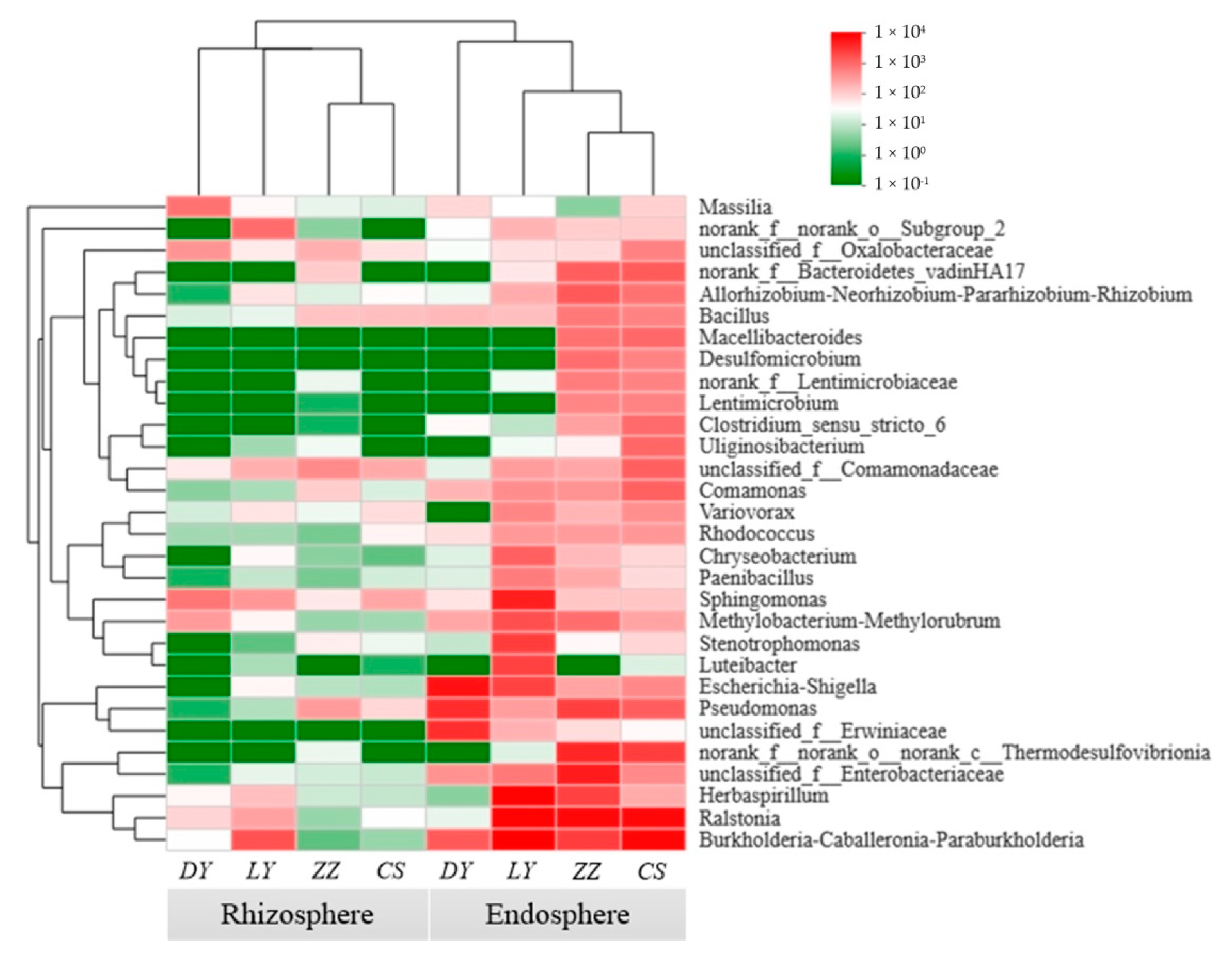
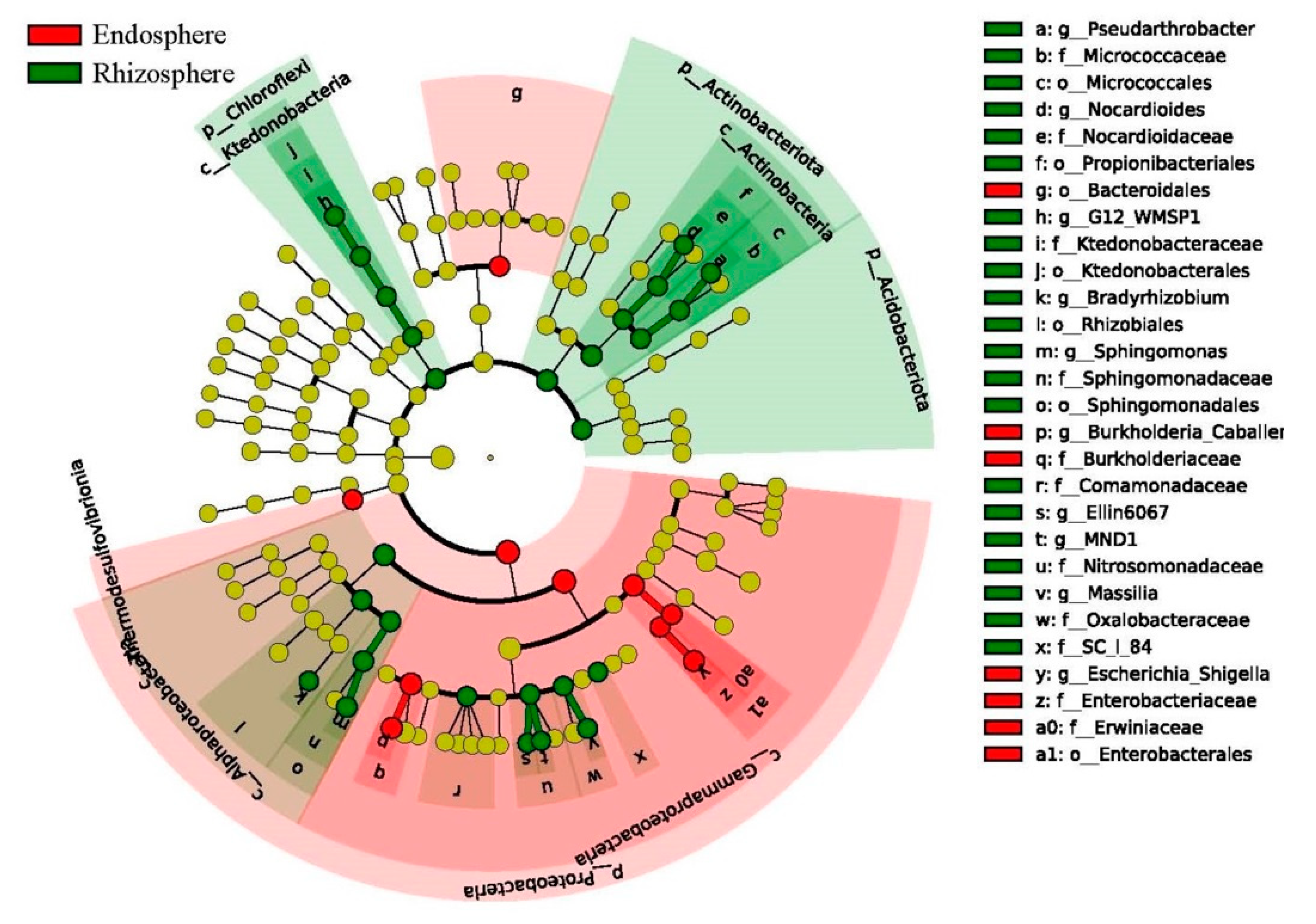
3.7. Discriminative Taxon between the Endosphere and Rhizosphere Bacteria Community
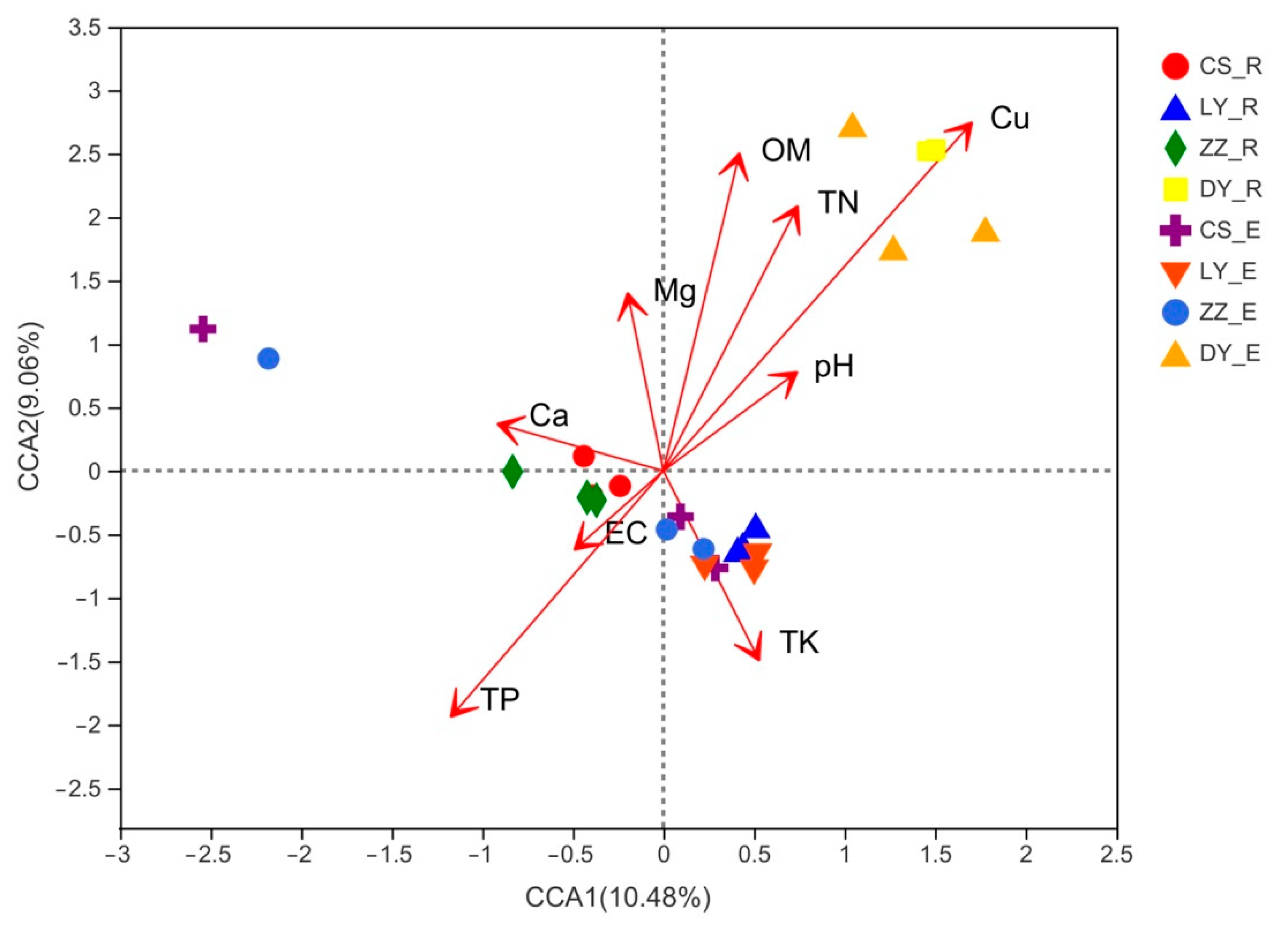
3.8. Relationships between Community Structure and Environmental Factors
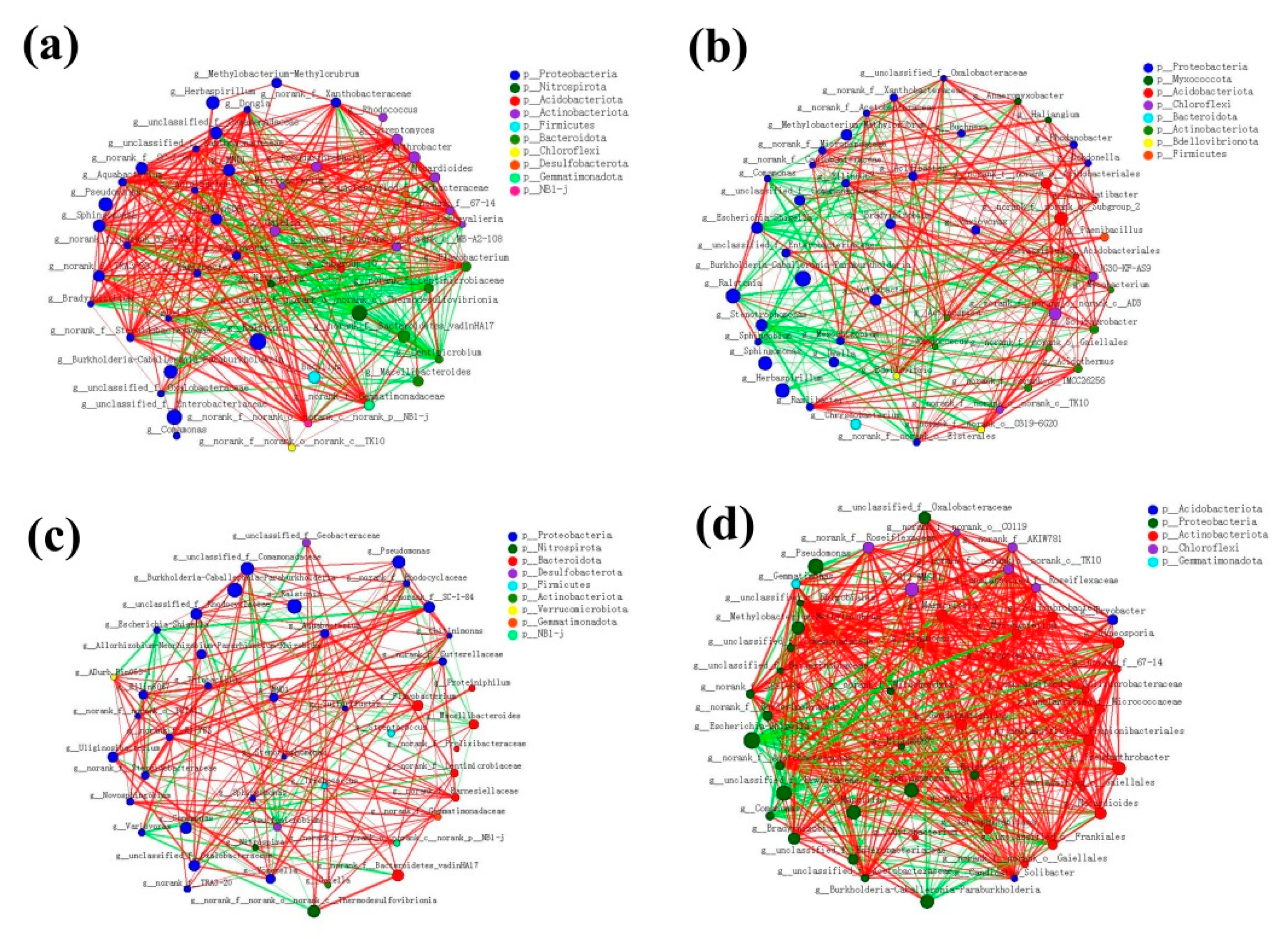
3.9. Molecular Ecological Network of the Bacterial Communities
4. Discussion
4.1. Variations of Bacterial Community Diversity and Structure between Rhizosphere and Endosphere
4.2. Factors Influencing Bacterial Community Diversity and Composition
4.3. The Dominant Genera in the Endosphere and Rhizosphere of C. communis under High Level Cu Contamination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, Q.; Li, Z.; Lu, X.; Duan, Q.; Huang, L.; Bi, J. A review of soil heavy metal pollution from industrial and agricultural regions in China: Pollution and risk assessment. Sci. Total Environ. 2018, 642, 690–700. [Google Scholar] [CrossRef]
- Gu, T.Y.; Yu, H.; Li, F.; Zeng, W.M.; Wu, X.L.; Shen, L.; Yu, R.L.; Liu, Y.D.; Li, J.K. Antimony-oxidizing bacteria alleviate Sb stress in Arabidopsis by attenuating Sb toxicity and reducing Sb uptake. Plant Soil 2020, 452, 397–412. [Google Scholar] [CrossRef]
- Liu, L.; Li, W.; Song, W.; Guo, M. Remediation techniques for heavy metal-contaminated soils: Principles and applicability. Sci. Total Environ. 2018, 633, 206–219. [Google Scholar] [CrossRef]
- Etesami, H.; Maheshwari, D.K. Use of plant growth promoting rhizobacteria (PGPRs) with multiple plant growth promoting traits in stress agriculture: Action mechanisms and future prospects. Ecotoxicol. Environ. Saf. 2018, 156, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Singh, R.; Arora, N.K. Alleviation of heavy metal stress in plants and remediation of soil by rhizosphere microorganisms. Front. Microbiol. 2017, 8, 1706. [Google Scholar] [CrossRef]
- Asad, S.A.; Farooq, M.; Afzal, A.; West, H. Integrated phytobial heavy metal remediation strategies for a sustainable clean environment—A review. Chemosphere 2019, 217, 925–941. [Google Scholar] [CrossRef] [PubMed]
- Manoj, S.R.; Karthik, C.; Kadirvelu, K.; Arulselvi, P.I.; Shanmugasundaram, T.; Bruno, B.; Rajkumar, M. Understanding the molecular mechanisms for the enhanced phytoremediation of heavy metals through plant growth promoting rhizobacteria: A review. J. Environ. Manag. 2020, 254, 109779. [Google Scholar] [CrossRef]
- Etesami, H. Bacterial mediated alleviation of heavy metal stress and decreased accumulation of metals in plant tissues: Mechanisms and future prospects. Ecotoxicol. Environ. Saf. 2018, 147, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Li, F.; Wu, C.; Yu, R.; Wu, X.; Shen, L.; Liu, Y.; Qiu, G.; Li, J. Role of extracellular polymeric substance (EPS) in toxicity response of soil bacteria Bacillus sp. S3 to multiple heavy metals. Bioprocess. Biosyst. Eng. 2020, 43, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Visioli, G.; D’Egidio, S.; Sanangelantoni, A.M. The bacterial rhizobiome of hyperaccumulators: Future perspectives based on omics analysis and advanced microscopy. Front. Plant Sci. 2014, 5, 752. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Hou, D.; Chen, J.; Li, J.; Fu, Y.; Wang, S.; Zheng, W.; Lu, L.; Tian, S. Distinct rhizobacterial functional assemblies assist two Sedum alfredii ecotypes to adopt different survival strategies under lead stress. Environ. Int. 2020, 143, 105912. [Google Scholar] [CrossRef]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellin, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voges, M.; Bai, Y.; Schulze-Lefert, P.; Sattely, E.S. Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 12558–12565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiong, W.; Fengshan, P.; Xiaomeng, X.; Rafiq, M.T.; Xiao’e, Y.; Bao, C.; Ying, F. Cadmium level and soil type played a selective role in the endophytic bacterial community of hyperaccumulator Sedum alfredii Hance. Chemosphere 2021, 263, 127986. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Chen, Q.L.; He, C.; Shi, Q.; Chen, S.C.; Reid, B.J.; Zhu, Y.G.; Sun, G.X. Organic carbon amendments affect the chemodiversity of soil dissolved organic matter and its associations with soil microbial communities. Environ. Sci. Technol. 2019, 53, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, W.; Tan, J.; Wang, Y.; Qin, Y.; He, H.; Wu, H.; Zuo, W.; He, D. Responses of the rhizosphere bacterial community in acidic crop soil to pH: Changes in diversity, composition, interaction, and function. Sci. Total Environ. 2020, 700, 134418. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, K.M.; Silver, W.L.; Thompson, A.W.; Firestone, M.K. Microbial communities acclimate to recurring changes in soil redox potential status. Environ. Microbiol. 2010, 12, 3137–3149. [Google Scholar] [CrossRef]
- Schneider, A.R.; Gommeaux, M.; Duclercq, J.; Fanin, N.; Conreux, A.; Alahmad, A.; Lacoux, J.; Roger, D.; Spicher, F.; Ponthieu, M.; et al. Response of bacterial communities to Pb smelter pollution in contrasting soils. Sci. Total Environ. 2017, 605-606, 436–444. [Google Scholar] [CrossRef]
- Li, J.; Xu, H.; Song, Y.; Tang, L.; Gong, Y.; Yu, R.; Shen, L.; Wu, X.; Liu, Y.; Zeng, W. Geography plays a more important role than soil composition on structuring genetic variation of pseudometallophyte Commelina communis. Front. Plant Sci. 2016, 7, 1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Liao, B.; Li, J.T.; Mengoni, A.; Hu, M.; Luo, W.C.; Shu, W.S. Contrasting patterns of genetic divergence in two sympatric pseudo-metallophytes: Rumex acetosa L. and Commelina communis L. BMC Evol. Biol. 2012, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- He, L.Y.; Zhang, Y.F.; Ma, H.Y.; Su, L.N.; Chen, Z.J.; Wang, Q.Y.; Qian, M.; Sheng, X.F. Characterization of copper-resistant bacteria and assessment of bacterial communities in rhizosphere soils of copper-tolerant plants. Appl. Soil Ecol. 2010, 44, 49–55. [Google Scholar] [CrossRef]
- Sun, L.N.; Zhang, Y.F.; He, L.Y.; Chen, Z.J.; Wang, Q.Y.; Qian, M.; Sheng, X.F. Genetic diversity and characterization of heavy metal-resistant-endophytic bacteria from two copper-tolerant plant species on copper mine wasteland. Bioresour. Technol. 2010, 101, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Bao, J.; Zheng, J.; Xu, F.; Wang, L. Phytoremediation of heavy metal contaminated soil potential by woody plants on Tonglushan ancient copper spoil heap in China. Int. J. Phytoremediation 2018, 20, 1–7. [Google Scholar] [CrossRef]
- Sun, X.; Zhou, Y.; Tan, Y.; Wu, Z.; Lu, P.; Zhang, G.; Yu, F. Restoration with pioneer plants changes soil properties and remodels the diversity and structure of bacterial communities in rhizosphere and bulk soil of copper mine tailings in Jiangxi Province, China. Environ. Sci. Pollut. Res. Int. 2018, 25, 22106–22119. [Google Scholar] [CrossRef]
- Lee, S.A.; Kim, Y.; Kim, J.M.; Chu, B.; Joa, J.H.; Sang, M.K.; Song, J.; Weon, H.Y. A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci. Rep. 2019, 9, 9300. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.J.; Wang, L.L.; Li, Q.; Shang, Q.M. Bacterial communities in the rhizosphere, phyllosphere and endosphere of tomato plants. PLoS ONE 2019, 14, e0223847. [Google Scholar] [CrossRef]
- Beckers, B.; Op De Beeck, M.; Thijs, S.; Truyens, S.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Performance of 16s rDNA primer pairs in the study of rhizosphere and endosphere bacterial microbiomes in metabarcoding studies. Front. Microbiol. 2016, 7, 650. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Ma, Q.; Chen, Y.; Tian, B.; Xu, L.; Bai, Y.; Chen, W.; Li, X. Variation in rhizosphere microbial communities and its association with the symbiotic efficiency of rhizobia in soybean. ISME J. 2020, 14, 1915–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Muller, D.B.; Srinivas, G.; Garrido-Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Munch, P.C.; Spaepen, S.; Remus-Emsermann, M.; et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Niu, B.; Paulson, J.N.; Zheng, X.; Kolter, R. Simplified and representative bacterial community of maize roots. Proc. Natl. Acad. Sci. USA 2017, 114, E2450–E2459. [Google Scholar] [CrossRef] [Green Version]
- Donn, S.; Kirkegaard, J.A.; Perera, G.; Richardson, A.E.; Watt, M. Evolution of bacterial communities in the wheat crop rhizosphere. Environ. Microbiol. 2015, 17, 610–621. [Google Scholar] [CrossRef]
- Lopez-Echartea, E.; Strejcek, M.; Mukherjee, S.; Uhlik, O.; Yrjala, K. Bacterial succession in oil-contaminated soil under phytoremediation with poplars. Chemosphere 2020, 243, 125242. [Google Scholar] [CrossRef]
- Huang, Y.H. Comparison of rhizosphere and endophytic microbial communities of Chinese leek through high-throughput 16S rRNA gene Illumina sequencing. J. Integr. Agric. 2018, 17, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Liu, C.; Li, B.; Dong, Y. Trifolium repens L. regulated phytoremediation of heavy metal contaminated soil by promoting soil enzyme activities and beneficial rhizosphere associated microorganisms. J. Hazard. Mater. 2021, 402, 123829. [Google Scholar] [CrossRef]
- Tian, X.Y.; Zhang, C.S. Illumina-based analysis of endophytic and rhizosphere bacterial diversity of the coastal halophyte Messerschmidia sibirica. Front. Microbiol. 2017, 8, 2288. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Robert, C.A.M.; Cadot, S.; Zhang, X.; Ye, M.; Li, B.; Manzo, D.; Chervet, N.; Steinger, T.; van der Heijden, M.G.A.; et al. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 2018, 9, 2738. [Google Scholar] [CrossRef] [Green Version]
- Canarini, A.; Kaiser, C.; Merchant, A.; Richter, A.; Wanek, W. Root exudation of primary metabolites: Mechanisms and their roles in plant responses to environmental stimuli. Front. Plant Sci. 2019, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gong, X. Niche differentiation rather than biogeography shapes the diversity and composition of microbiome of Cycas panzhihuaensis. Microbiome 2019, 7, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckers, B.; Op De Beeck, M.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Xiang, X.; Ma, L.; Evers, S.; Wang, R.; Qiu, X.; Wang, H. Rare species shift the structure of bacterial communities across Sphagnum compartments in a subalpine peatland. Front. Microbiol. 2019, 10, 3138. [Google Scholar] [CrossRef]
- Coleman-Derr, D.; Desgarennes, D.; Fonseca-Garcia, C.; Gross, S.; Clingenpeel, S.; Woyke, T.; North, G.; Visel, A.; Partida-Martinez, L.P.; Tringe, S.G. Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 2016, 209, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Chen, P.; Wang, X.; Li, H.; Zuo, L.; Zhang, Y.; Li, L. Structural variability and niche differentiation of the rhizosphere and endosphere fungal microbiome of Casuarina equisetifolia at different ages. Braz. J. Microbiol. 2020, 51, 1873–1884. [Google Scholar] [CrossRef]
- Cregger, M.A.; Veach, A.M.; Yang, Z.K.; Crouch, M.J.; Vilgalys, R.; Tuskan, G.A.; Schadt, C.W. The Populus holobiont: Dissecting the effects of plant niches and genotype on the microbiome. Microbiome 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Zhang, S.; Zhong, Q.; Gong, G.; Wang, G.; Guo, X.; Xu, X. Effects of soil chemical properties and fractions of Pb, Cd, and Zn on bacterial and fungal communities. Sci. Total Environ. 2020, 715, 136904. [Google Scholar] [CrossRef] [PubMed]
- Chodak, M.; Gołębiewski, M.; Morawska-Płoskonka, J.; Kuduk, K.; Niklińska, M. Diversity of microorganisms from forest soils differently polluted with heavy metals. Appl. Soil Ecol. 2013, 64, 7–14. [Google Scholar] [CrossRef]
- Yu, H.; Zheng, X.; Weng, W.; Yan, X.; Chen, P.; Liu, X.; Peng, T.; Zhong, Q.; Xu, K.; Wang, C.; et al. Synergistic effects of antimony and arsenic contaminations on bacterial, archaeal and fungal communities in the rhizosphere of Miscanthus sinensis: Insights for nitrification and carbon mineralization. J. Hazard. Mater. 2021, 411, 125094. [Google Scholar] [CrossRef]
- Jiang, B.; Adebayo, A.; Jia, J.; Xing, Y.; Deng, S.; Guo, L.; Liang, Y.; Zhang, D. Impacts of heavy metals and soil properties at a Nigerian e-waste site on soil microbial community. J. Hazard. Mater. 2019, 362, 187–195. [Google Scholar] [CrossRef]
- Li, X.; Meng, D.; Li, J.; Yin, H.; Liu, H.; Liu, X.; Cheng, C.; Xiao, Y.; Liu, Z.; Yan, M. Response of soil microbial communities and microbial interactions to long-term heavy metal contamination. Environ. Pollut. 2017, 231, 908–917. [Google Scholar] [CrossRef]
- Guo, H.; Nasir, M.; Lv, J.; Dai, Y.; Gao, J. Understanding the variation of microbial community in heavy metals contaminated soil using high throughput sequencing. Ecotoxicol. Environ. Saf. 2017, 144, 300–306. [Google Scholar] [CrossRef]
- Bastida, F.; Selevsek, N.; Torres, I.F.; Hernandez, T.; Garcia, C. Soil restoration with organic amendments: Linking cellular functionality and ecosystem processes. Sci. Rep. 2015, 5, 15550. [Google Scholar] [CrossRef] [Green Version]
- Benidire, L.; Pereira, S.I.A.; Naylo, A.; Castro, P.M.L.; Boularbah, A. Do metal contamination and plant species affect microbial abundance and bacterial diversity in the rhizosphere of metallophytes growing in mining areas in a semiarid climate? J. Soil Sediment. 2019, 20, 1003–1017. [Google Scholar] [CrossRef]
- Liu, Z.F.; Fu, B.J.; Zheng, X.X.; Liu, G.H. Plant biomass, soil water content and soil N:P ratio regulating soil microbial functional diversity in a temperate steppe: A regional scale study. Soil Biol. Biochem. 2010, 42, 445–450. [Google Scholar] [CrossRef]
- Shen, C.C.; Xiong, J.B.; Zhang, H.Y.; Feng, Y.Z.; Lin, X.G.; Li, X.Y.; Liang, W.J.; Chu, H.Y. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol. Biochem. 2013, 57, 204–211. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Zhao, Z.; Gong, Q.; Zhai, B.; Li, Z. Responses of fungal–bacterial community and network to organic inputs vary among different spatial habitats in soil. Soil Biol. Biochem. 2018, 125, 54–63. [Google Scholar] [CrossRef]
- Zhang, X.M.; He, R.J.; Su, R.; Zeng, J.; Zhou, Q.; Huang, R.; Zhao, D.Y.; Guo, L.; He, F.; Yu, Z.B. Composition and co-occurrence network of the rhizosphere bacterial community of two emergent macrophytes and implications for phytoremediation. Mar. Freshwater Res. 2021, 72, 1053–1064. [Google Scholar] [CrossRef]
- Sheng, X.; Sun, L.; Huang, Z.; He, L.; Zhang, W.; Chen, Z. Promotion of growth and Cu accumulation of bio-energy crop (Zea mays) by bacteria: Implications for energy plant biomass production and phytoremediation. J. Environ. Manag. 2012, 103, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, Y.; Rafiq, M.T.; Khan, K.Y.; Pan, F.; Yang, X.; Feng, Y. Improvement of cadmium uptake and accumulation in Sedum alfredii by endophytic bacteria Sphingomonas SaMR12: Effects on plant growth and root exudates. Chemosphere 2014, 117, 367–373. [Google Scholar] [CrossRef]
- Pan, F.; Meng, Q.; Luo, S.; Shen, J.; Chen, B.; Khan, K.Y.; Japenga, J.; Ma, X.; Yang, X.; Feng, Y. Enhanced Cd extraction of oilseed rape (Brassica napus) by plant growth-promoting bacteria isolated from Cd hyperaccumulator Sedum alfredii Hance. Int. J. Phytoremediation 2017, 19, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, M.; Bruno, L.B.; Banu, J.R. Alleviation of environmental stress in plants: The role of beneficial Pseudomonas spp. Crit. Rev. Environ. Sci. Technol. 2017, 47, 372–407. [Google Scholar] [CrossRef]
- Xu, C.; Chen, X.; Duan, D.; Peng, C.; Le, T.; Shi, J. Effect of heavy-metal-resistant bacteria on enhanced metal uptake and translocation of the Cu-tolerant plant, Elsholtzia splendens. Environ. Sci. Pollut. Res. Int. 2015, 22, 5070–5081. [Google Scholar] [CrossRef]
- Babu, A.G.; Shea, P.J.; Sudhakar, D.; Jung, I.B.; Oh, B.T. Potential use of Pseudomonas koreensis AGB-1 in association with Miscanthus sinensis to remediate heavy metal(loid)-contaminated mining site soil. J. Environ. Manag. 2015, 151, 160–166. [Google Scholar] [CrossRef]
- Sun, W.; Xiong, Z.; Chu, L.; Li, W.; Soares, M.A.; White, J.F., Jr.; Li, H. Bacterial communities of three plant species from Pb-Zn contaminated sites and plant-growth promotional benefits of endophytic Microbacterium sp. (strain BXGe71). J. Hazard. Mater. 2019, 370, 225–231. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Ren, Y.; Zeng, W.; Wu, X.; Shen, L.; Yu, R.; Liu, Y.; Li, J. Deciphering the Endophytic and Rhizospheric Microbial Communities of a Metallophyte Commelina communis in Different Cu-Polluted Soils. Microorganisms 2021, 9, 1689. https://doi.org/10.3390/microorganisms9081689
He L, Ren Y, Zeng W, Wu X, Shen L, Yu R, Liu Y, Li J. Deciphering the Endophytic and Rhizospheric Microbial Communities of a Metallophyte Commelina communis in Different Cu-Polluted Soils. Microorganisms. 2021; 9(8):1689. https://doi.org/10.3390/microorganisms9081689
Chicago/Turabian StyleHe, Li, Yanzhen Ren, Weimin Zeng, Xueling Wu, Li Shen, Runlan Yu, Yuandong Liu, and Jiaokun Li. 2021. "Deciphering the Endophytic and Rhizospheric Microbial Communities of a Metallophyte Commelina communis in Different Cu-Polluted Soils" Microorganisms 9, no. 8: 1689. https://doi.org/10.3390/microorganisms9081689
APA StyleHe, L., Ren, Y., Zeng, W., Wu, X., Shen, L., Yu, R., Liu, Y., & Li, J. (2021). Deciphering the Endophytic and Rhizospheric Microbial Communities of a Metallophyte Commelina communis in Different Cu-Polluted Soils. Microorganisms, 9(8), 1689. https://doi.org/10.3390/microorganisms9081689