Identification and Characterization of Hypothalamic Alternative Splicing Events and Variants in Ovine Fecundity-Related Genes

 and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Selection and Samples Preparation
2.2. Total RNA Extraction and Sequencing
2.3. Filtering Raw Data and Mapping
2.4. Transcriptome Assembly and Differential Expression Analysis
2.5. Identification of AS Events, SNPs and Indels
2.6. Correlation Analysis between Proteomics and Transcriptomics
2.7. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
3. Results
3.1. mRNAs Profiling
3.2. Identification and Characterization of AS Events
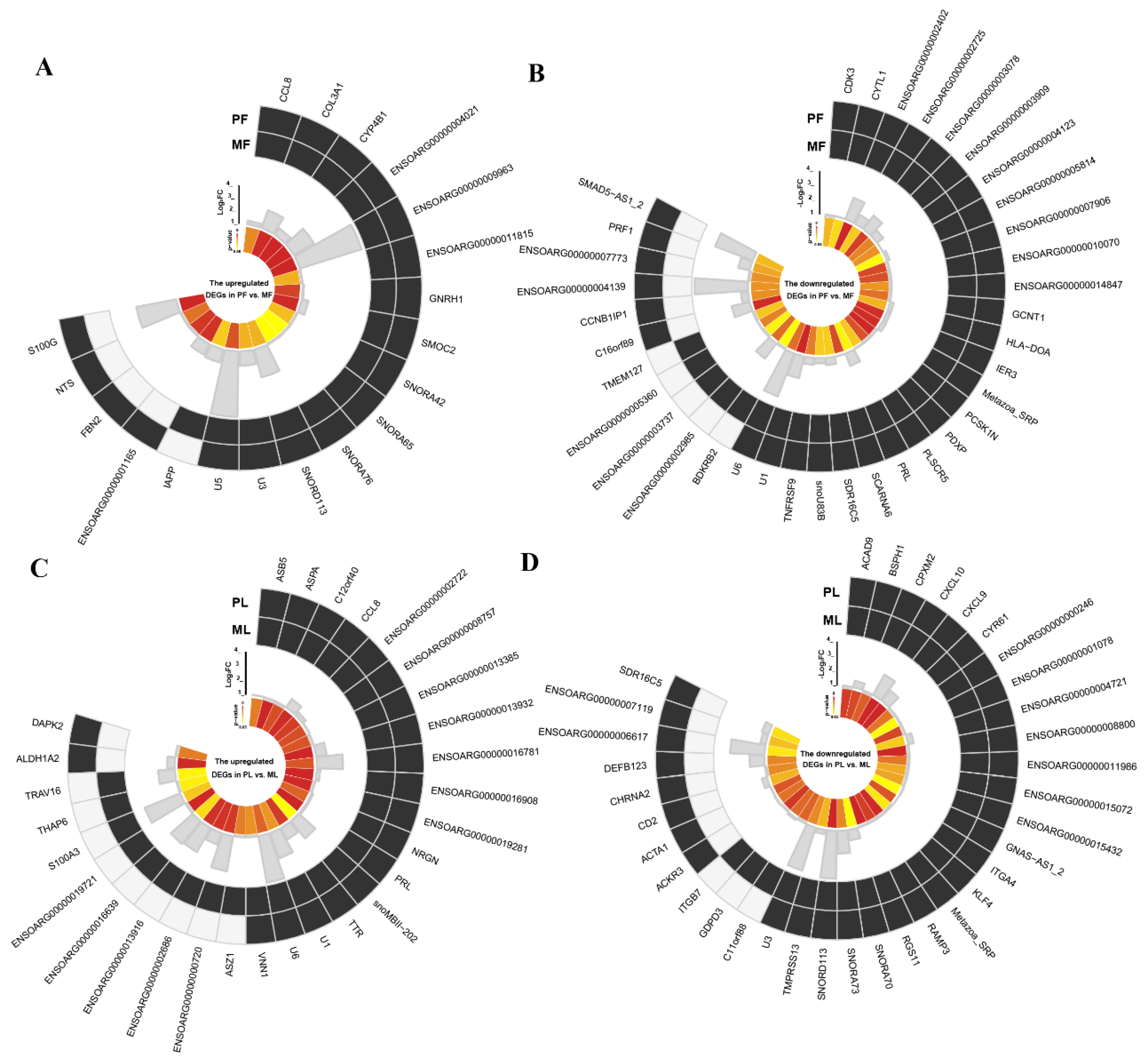
3.3. The AS Events Found in DEGs
3.4. Functional Enrichment Analysis of DE AS Genes
3.5. Interaction Analysis of DE AS Genes with microRNA (miRNA)
3.6. The SNPs and Indels Analysis of DEGs
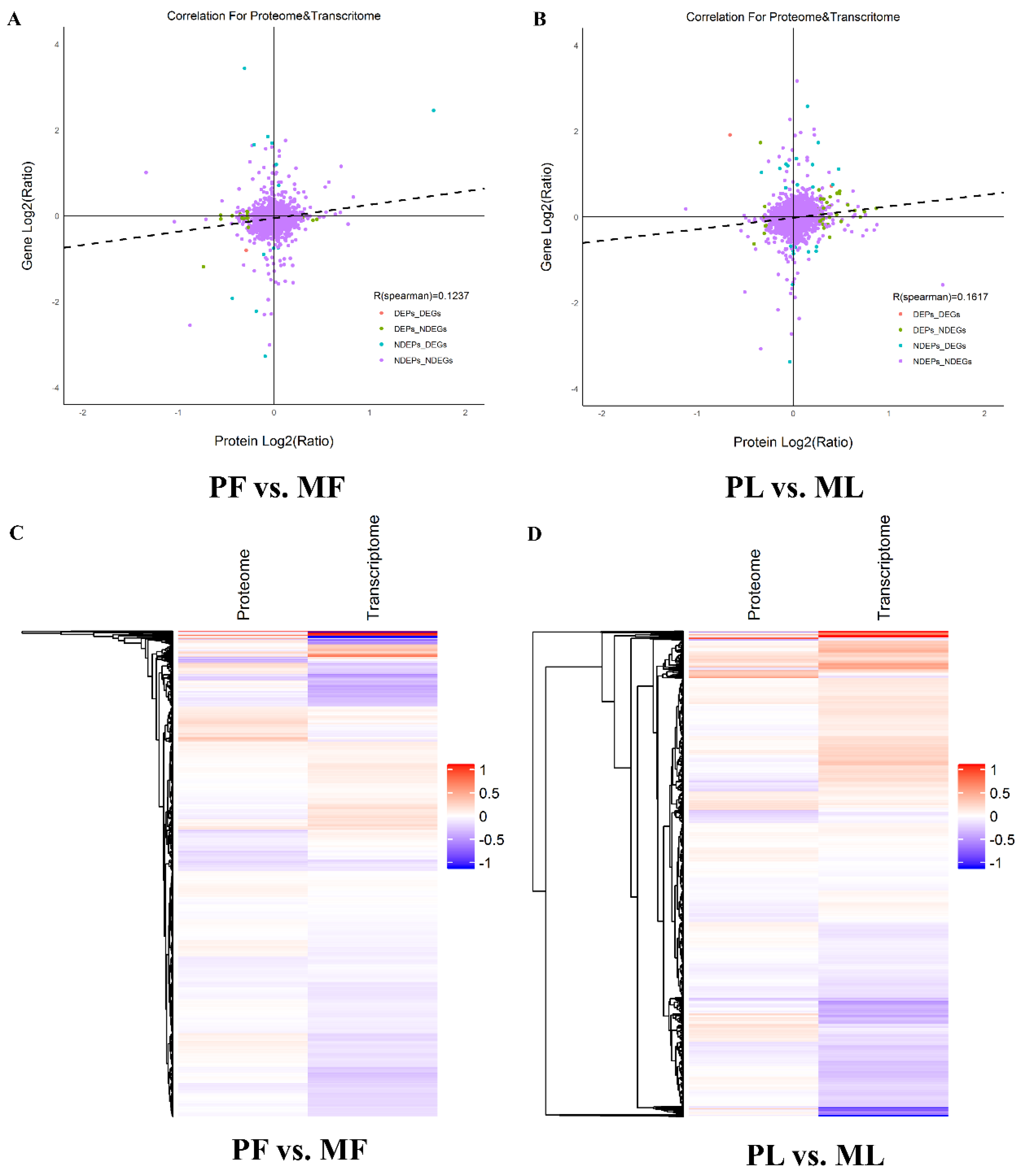
3.7. Correlation Analysis of the Proteomics and Transcriptomics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isken, O.; Maquat, L.E. The multiple lives of NMD factors: Balancing roles in gene and genome regulation. Nat. Rev. Genet. 2008, 9, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.M.; Castle, J.; Garrett-Engele, P.; Kan, Z.; Loerch, P.M.; Armour, C.D.; Santos, R.; Schadt, E.E.; Stoughton, R.; Shoemaker, D.D. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003, 302, 2141–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Elisabetta, F.; Scheiffele, P. Regulation of neuronal differentiation, function, and plasticity by alternative splicing. Annu. Rev. Cell Dev. Biol. 2018, 34, 451–469. [Google Scholar]
- Liu, Q.; Wang, X.; Kong, X.; Yang, X.; Cheng, R.; Zhang, W.; Gao, P.; Chen, L.; Wang, Z.; Fang, Y.; et al. Prognostic alternative mRNA splicing signature and a novel biomarker in triple-negative breast cancer. DNA Cell Biol. 2020, 39, 1051–1063. [Google Scholar] [CrossRef]
- Guo, Z.; Zhu, H.; Xu, W.; Wang, X.; Liu, H.; Wu, Y.; Wang, M.; Chu, H.; Zhang, Z. Alternative splicing related genetic variants contribute to bladder cancer risk. Mol. Carcinog. 2020. [Google Scholar] [CrossRef]
- Li, B.; He, X.; Zhao, Y.; Bai, D.; Du, M.; Song, L.; Liu, Z.; Yin, Z.; Manglai, D. Transcriptome profiling of developing testes and spermatogenesis in the Mongolian horse. BMC Genet. 2020, 21, 46. [Google Scholar] [CrossRef]
- Chen, M.; Yang, W.; Liu, N.; Zhang, X.; Dong, W.; Lan, X.; Pan, C. Pig Hsd17b3: Alternative splice variants expression, insertion/deletion (indel) in promoter region and their associations with male reproductive traits. J. Steroid Biochem. Mol. Biol. 2019, 195, 105483. [Google Scholar] [CrossRef] [PubMed]
- Anamthathmakula, P.; Kyathanahalli, C.; Ingles, J.; Hassan, S.S.; Condon, J.C.; Jeyasuria, P. Estrogen receptor alpha isoform ERdelta7 in myometrium modulates uterine quiescence during pregnancy. EBioMedicine 2019, 39, 520–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Ovarian transcriptomic analysis reveals the alternative splicing events associated with fecundity in different sheep breeds. Anim. Reprod. Sci. 2018, 198, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, N.M. A review of the effects of the Booroola gene (FecB) on sheep production. Small Rumin. Res. 2009, 85, 75–84. [Google Scholar] [CrossRef]
- Liu, Q.; Pan, Z.; Wang, X.; Hu, W.; Di, R.; Yao, Y.; Chu, M. Progress on major genes for high fecundity in ewes. Front. Agric. Sci. Eng. 2014, 1, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Våge, D.I.; Husdal, M.; Kent, M.P.; Klemetsdal, G.; Boman, I.A. A missense mutation in growth differentiation factor 9 (GDF9) is strongly associated with litter size in sheep. BMC Genet. 2013, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Calvo, J.H.; Chantepie, L.; Serrano, M.; Sarto, M.P.; Iguacel, L.P.; Jiménez, M.Á.; Alabart, J.L.; Folch, J.; Fabre, S.; Lahoz, B. A new allele in the BMP15 gene (FecX) that affects prolificacy co-segregates with FecX and FecX in Rasa aragonesa sheep. Theriogenology 2020, 144, 107–111. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Q.; Di, R.; Hu, W.; Wang, X.; He, X.; Ma, L.; Chu, M. Single nucleotide polymorphisms in BMP2 and BMP7 and the association with litter size in Small Tail Han sheep. Anim. Reprod. Sci. 2019, 204, 183–192. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; He, X.; Di, R.; Chu, M. Mutations in NLRP5 and NLRP9 are associated with litter size in Small Tail Han sheep. Animals 2020, 10, 689. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Bhagwate, A.; Prodduturi, N.; Yang, P.; Kocher, J.A. Indel detection from RNA-seq data: Tool evaluation and strategies for accurate detection of actionable mutations. Briefings Bioinform. 2017, 18, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Hui, Y.; Zhang, Y.; Wang, K.; Pan, C.; Chen, H.; Qu, L.; Song, X.; Lan, X. Goat DNMT3B: An indel mutation detection, association analysis with litter size and mRNA expression in gonads. Theriogenology 2020, 147, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Kang, Z.; Jiang, E.; Yan, H.; Zhu, H.; Liu, J.; Qu, L.; Lan, X.; Pan, C. Genetic effects of DSCAML1 identified in genome-wide association study revealing strong associations with litter size and semen quality in goat (Capra hircus). Theriogenology 2020, 146, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Lu, T.; Zhao, Z.; Liu, G.; Lian, Z.; Guo, Y.; Sun, B.; Liu, D.; Li, Y. Comprehensive analysis of mRNAs and miRNAs in the ovarian follicles of uniparous and multiple goats at estrus phase. BMC Genom. 2020, 21, 267. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wang, Z.; Yang, H.; Yao, X.; Yang, P.; Ren, C.; Wang, F.; Zhang, Y. Pituitary transcriptomic study reveals the differential regulation of lncRNAs and mRNAs related to prolificacy in different FecB genotyping sheep. Genes 2019, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tang, J.; Di, R.; Liu, Q.; Wang, X.; Gan, S.; Zhang, X.; Zhang, J.; Chu, M.; Hu, W. Integrated hypothalamic transcriptome profiling reveals the reproductive roles of mRNAs and miRNAs in sheep. Front. Genet. 2019, 10, 1296. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Hu, W.; Chen, S.; Di, R.; Liu, Q.; Wang, X.; He, X.; Gan, S.; Zhang, X.; Zhang, J.; et al. The genetic mechanism of high prolificacy in small tail han sheep by comparative proteomics of ovaries in the follicular and luteal stages. J. Proteom. 2019, 204, 103394. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; Di, R.; Liu, Q.; Wang, X.; Gan, S.; Zhang, X.; Zhang, J.; Chen, W.; Hu, W.; et al. Identification of prolificacy-related differentially expressed proteins from sheep (Ovis aries) hypothalamus by comparative proteomics. Proteomics 2019, 19, e1900118. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, W.; He, X.; Pan, Z.; Guo, X.; Feng, T.; Cao, G.; Huang, D.; He, J.; Di, R.; et al. Establishment of high-throughput molecular detection methods for ovine high fecundity major gene and their application. Acta Veteinaria Zootech. Sin. 2017, 48, 39–51. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res 2013, 2, 188. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wu, P.; Yang, Z.; Deng, S.; Ni, L.; Zhang, Y.; Jin, L.; Pan, Y. miR-193a-5p promotes pancreatic cancer cell metastasis through SRSF6-mediated alternative splicing of OGDHL and ECM1. Am. J. Cancer Res. 2020, 10, 38–59. [Google Scholar]
- Popovitchenko, T.; Park, Y.; Page, N.F.; Luo, X.; Krsnik, Z.; Liu, Y.; Salamon, I.; Stephenson, J.D.; Kraushar, M.L.; Volk, N.L.; et al. Translational derepression of Elavl4 isoforms at their alternative 5’ UTRs determines neuronal development. Nat. Commun. 2020, 11, 1674. [Google Scholar] [CrossRef]
- Gopalakrishnan, K.; Kumar, S. Whole-genome uterine artery transcriptome profiling and alternative splicing analysis in rat pregnancy. Int. J. Mol. Sci. 2020, 21, 2079. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Montes, A.M.; Fernández, A.; Pérez-Montarelo, D.; Alves, E.; Benítez, R.M.; Nuez, Y.; Óvilo, C.; Ibaez-Escriche, N.; Folch, J.M.; Fernández, A.I. Using RNA-Seq SNP data to reveal potential causal mutations related to pig production traits and RNA editing. Anim. Genet. 2017, 48, 151–165. [Google Scholar] [CrossRef]
- Fischer, D.; Laiho, A.; Gyenesei, A.; Sironen, A. Identification of reproduction-related gene polymorphisms using whole transcriptome sequencing in the large white pig population. G3 2015, 5, 1351–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultan, M.; Amstislavskiy, V.; Risch, T.; Schuette, M.; Dökel, S.; Ralser, M.; Balzereit, D.; Lehrach, H.; Yaspo, M.L. Influence of RNA extraction methods and library selection schemes on RNA-seq data. BMC Genom. 2014, 15, 675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, H.A.; Shupnik, M.A. Mechanisms for pulsatile regulation of the gonadotropin subunit genes by GNRH1. Biol. Reprod. 2006, 74, 993–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, T.J.; Hahn, T.P.; MacDougall-Shackleton, S.A.; Ball, G.F. Gonadotropin-releasing hormone plasticity: A comparative perspective. Front. Neuroendocr. 2012, 33, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Xiao, J.; Fan, Y.; Yang, K.; Li, K.; Wang, X.; Lu, Y.; Zhou, Y. miR-29 family regulates the puberty onset mediated by a novel Gnrh1 transcription factor TBX21. J. Endocrinol. 2019, 242, 185–197. [Google Scholar] [CrossRef] [PubMed]
- An, X.P.; Hou, J.X.; Zhao, H.B.; Li, G.; Bai, L.; Peng, J.Y.; Yan, Q.M.; Song, Y.X.; Wang, J.G.; Cao, B.Y. Polymorphism identification in goat GNRH1 and GDF9 genes and their association analysis with litter size. Anim. Genet. 2013, 44, 234–238. [Google Scholar] [CrossRef]
- Ko, E.K.; Chorich, L.P.; Sullivan, M.E.; Cameron, R.S.; Layman, L.C. JAK/STAT signaling pathway gene expression is reduced following Nelf knockdown in GnRH neurons. Mol. Cell. Endocrinol. 2018, 470, 151–159. [Google Scholar] [CrossRef]
- Bar-Lev, T.H.; Harris, D.; Tomić, M.; Stojilkovic, S.; Blumenfeld, Z.; Brown, P.; Seger, R.; Naor, Z. Role of PI4K and PI3K-AKT in ERK1/2 activation by GnRH in the pituitary gonadotropes. Mol. Cell. Endocrinol. 2015, 415, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Araujo-Lopes, R.; Crampton, J.R.; Aquino, N.S.; Miranda, R.M.; Kokay, I.C.; Reis, A.M.; Franci, C.R.; Grattan, D.R.; Szawka, R.E. Prolactin regulates kisspeptin neurons in the arcuate nucleus to suppress LH secretion in female rats. Endocrinology 2014, 155, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Lomet, D.; Druart, X.; Hazlerigg, D.; Beltramo, M.; Dardente, H. Circuit-level analysis identifies target genes of sex steroids in Ewe seasonal breeding. Mol. Cell. Endocrinol. 2020, 512, 110825. [Google Scholar] [CrossRef]
- Yoo, H.I.; Kim, E.G.; Lee, E.J.; Hong, S.Y.; Yoon, C.S.; Hong, M.J.; Park, S.J.; Woo, R.S.; Baik, T.K.; Song, D.Y. Neuroanatomical distribution of galectin-3 in the adult rat brain. J. Mol. Histol. 2017, 48, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Heinrichs, S.C.; Koob, G.F. Corticotropin-releasing factor in brain: A role in activation, arousal, and affect regulation. J. Pharmacol. Exp. Ther. 2004, 311, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Surendran, S.; Szucs, S.; Tyring, S.K.; Matalon, R. Aspartoacylase gene knockout in the mouse: Impact on reproduction. Reprod. Toxicol. 2005, 20, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Quintela, T.; Gonçalves, I.; Martinho, A.; Alves, C.H.; Saraiva, M.J.; Rocha, P.; Santos, C.R. Progesterone enhances transthyretin expression in the rat choroid plexus in vitro and in vivo via progesterone receptor. J. Mol. Neurosci. 2011, 44, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.; Gomes, J.R.; Saraiva, M.J. Transthyretin Induces insulin-like growth factor I nuclear translocation regulating its levels in the hippocampus. Mol. Neurobiol. 2015, 51, 1468–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiney, J.K.; Srivastava, V.K.; Pine, M.D.; Les Dees, W. Insulin-like growth factor-I activates KiSS-1 gene expression in the brain of the prepubertal female rat. Endocrinology 2009, 150, 376–384. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Base | Raw Reads | Clean Base | Clean Reads | Q30 (%) | Mapping Rate (%) |
|---|---|---|---|---|---|---|
| PF_H1 | 19,170,842,700 | 127,805,618 | 18,381,864,300 | 122,545,762 | 91.65 | 91.76 |
| PF_H2 | 18,659,879,100 | 124,399,194 | 17,994,065,700 | 119,960,438 | 93.22 | 92.80 |
| PF_H3 | 19,849,439,400 | 132,329,596 | 18,974,757,300 | 126,498,382 | 91.99 | 92.22 |
| PL_H1 | 19,326,903,300 | 128,846,022 | 18,789,727,800 | 125,264,852 | 93.80 | 92.81 |
| PL_H2 | 15,196,164,900 | 101,307,766 | 14,538,760,200 | 96,925,068 | 92.87 | 92.99 |
| PL_H3 | 20,028,519,600 | 133,523,464 | 19,408,391,100 | 129,389,274 | 93.78 | 92.78 |
| MF_H1 | 18,753,972,900 | 125,026,486 | 18,088,311,900 | 120,588,746 | 93.91 | 92.45 |
| MF_H2 | 19,560,182,400 | 130,401,216 | 18,550,470,900 | 123,669,806 | 92.84 | 92.40 |
| MF_H3 | 19,950,184,500 | 133,001,230 | 19,076,879,400 | 127,179,196 | 93.80 | 92.23 |
| ML_H1 | 19,028,904,900 | 126,859,366 | 18,356,811,000 | 122,378,740 | 94.39 | 92.77 |
| ML_H2 | 19,034,574,000 | 126,897,160 | 18,236,273,700 | 121,575,158 | 93.83 | 92.67 |
| ML_H3 | 19,262,332,500 | 128,415,550 | 18,641,870,100 | 124,279,134 | 94.15 | 92.20 |
| AS_type | PF_H1 | PF_H2 | PF_H3 | PL_H1 | PL_H2 | PL_H3 | MF_H1 | MF_H2 | MF_H3 | ML_H1 | ML_H2 | ML_H3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AE | 3.76% | 3.34% | 3.46% | 3.50% | 3.38% | 3.54% | 3.58% | 3.63% | 3.89% | 3.44% | 3.62% | 3.78% |
| XAE | 2.90% | 3.05% | 3.07% | 2.70% | 2.72% | 2.78% | 3.12% | 3.10% | 2.84% | 3.07% | 3.10% | 2.77% |
| IR | 7.15% | 7.70% | 7.28% | 6.46% | 7.04% | 6.66% | 7.68% | 7.71% | 6.47% | 7.47% | 7.36% | 6.52% |
| MIR | 1.72% | 2.20% | 1.99% | 1.55% | 1.76% | 1.71% | 2.19% | 2.12% | 1.66% | 2.08% | 2.18% | 1.66% |
| XIR | 2.66% | 3.02% | 2.83% | 2.64% | 2.67% | 2.64% | 3.04% | 3.10% | 2.38% | 3.31% | 2.96% | 2.52% |
| TTS | 32.80% | 33.00% | 33.19% | 33.80% | 34.09% | 33.59% | 32.88% | 32.40% | 32.99% | 33.08% | 32.72% | 33.59% |
| XMIR | 0.44% | 0.57% | 0.51% | 0.35% | 0.44% | 0.43% | 0.47% | 0.66% | 0.38% | 0.54% | 0.48% | 0.44% |
| SKIP | 7.39% | 6.10% | 6.47% | 6.89% | 6.47% | 6.96% | 6.51% | 6.73% | 7.62% | 6.21% | 6.88% | 7.25% |
| MSKIP | 1.33% | 1.06% | 1.19% | 1.31% | 1.12% | 1.33% | 1.12% | 1.26% | 1.56% | 1.14% | 1.21% | 1.36% |
| XMSKIP | 0.44% | 0.39% | 0.33% | 0.41% | 0.29% | 0.39% | 0.38% | 0.43% | 0.49% | 0.39% | 0.41% | 0.37% |
| XSKIP | 2.11% | 1.91% | 1.89% | 2.03% | 1.67% | 2.03% | 1.78% | 1.98% | 2.17% | 1.86% | 2.02% | 2.14% |
| TSS | 37.33% | 37.66% | 37.78% | 38.35% | 38.35% | 37.94% | 37.24% | 36.87% | 37.57% | 37.41% | 37.06% | 37.60% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Tang, J.; He, X.; Di, R.; Zhang, X.; Zhang, J.; Hu, W.; Chu, M. Identification and Characterization of Hypothalamic Alternative Splicing Events and Variants in Ovine Fecundity-Related Genes. Animals 2020, 10, 2111. https://doi.org/10.3390/ani10112111
Zhang Z, Tang J, He X, Di R, Zhang X, Zhang J, Hu W, Chu M. Identification and Characterization of Hypothalamic Alternative Splicing Events and Variants in Ovine Fecundity-Related Genes. Animals. 2020; 10(11):2111. https://doi.org/10.3390/ani10112111
Chicago/Turabian StyleZhang, Zhuangbiao, Jishun Tang, Xiaoyun He, Ran Di, Xiaosheng Zhang, Jinlong Zhang, Wenping Hu, and Mingxing Chu. 2020. "Identification and Characterization of Hypothalamic Alternative Splicing Events and Variants in Ovine Fecundity-Related Genes" Animals 10, no. 11: 2111. https://doi.org/10.3390/ani10112111
APA StyleZhang, Z., Tang, J., He, X., Di, R., Zhang, X., Zhang, J., Hu, W., & Chu, M. (2020). Identification and Characterization of Hypothalamic Alternative Splicing Events and Variants in Ovine Fecundity-Related Genes. Animals, 10(11), 2111. https://doi.org/10.3390/ani10112111