Comparative Transcriptomics Analysis of Testicular miRNA from Cryptorchid and Normal Horses

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
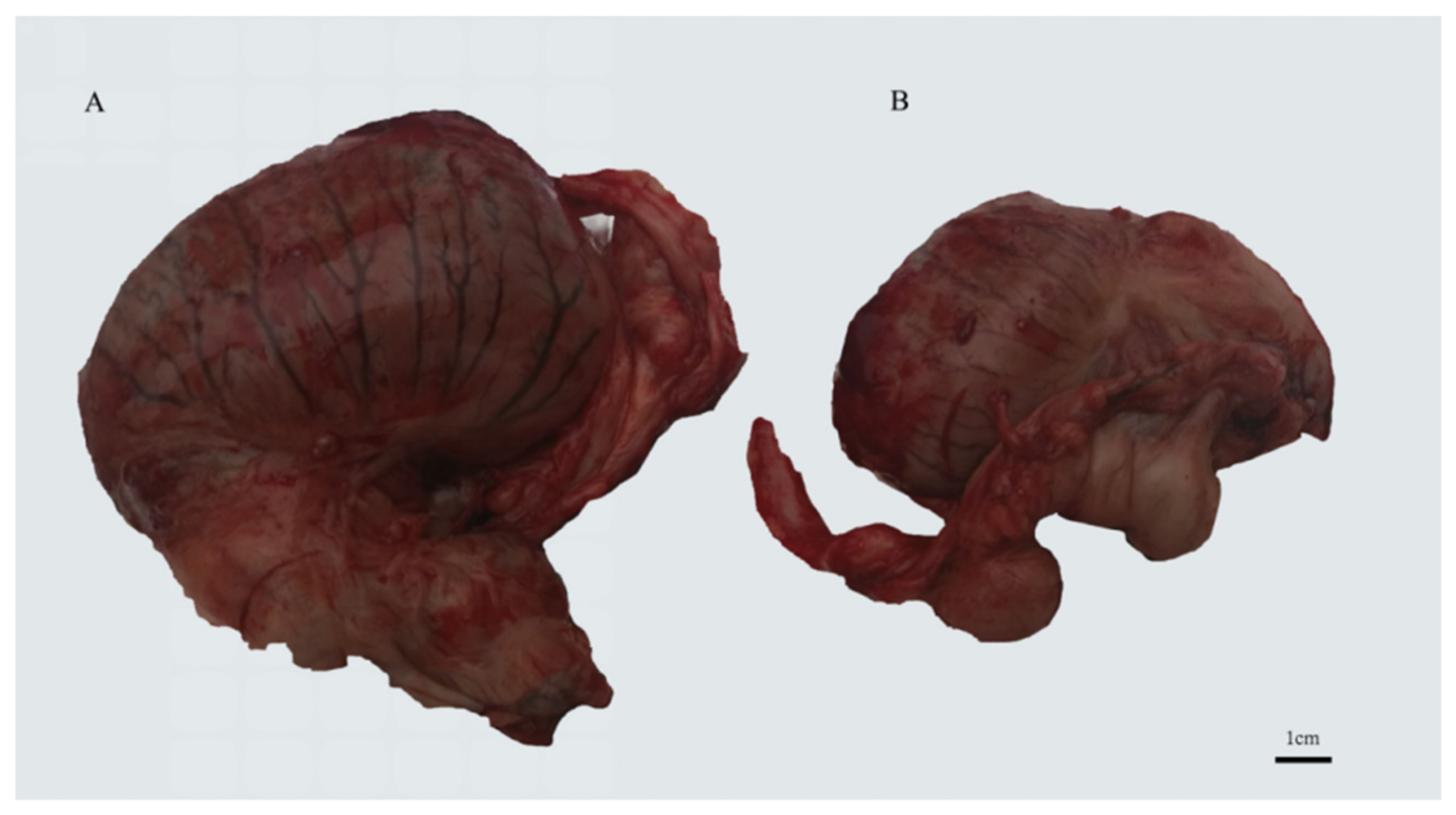
2.1. Horse Testis Tissue Collection
2.2. RNA Extraction and Quality Analysis
2.3. Small RNA Sequencing and Data Analysis
2.3.1. Library Preparation and Small RNA Sequencing
2.3.2. Read Filtering and Read Mapping on the Equine Reference Genome
2.3.3. Differential Expression Analysis of miRNAs
2.3.4. Validation of miRNA Expression by Quantitative Real-Time PCR (qRT-PCR)
3. Results
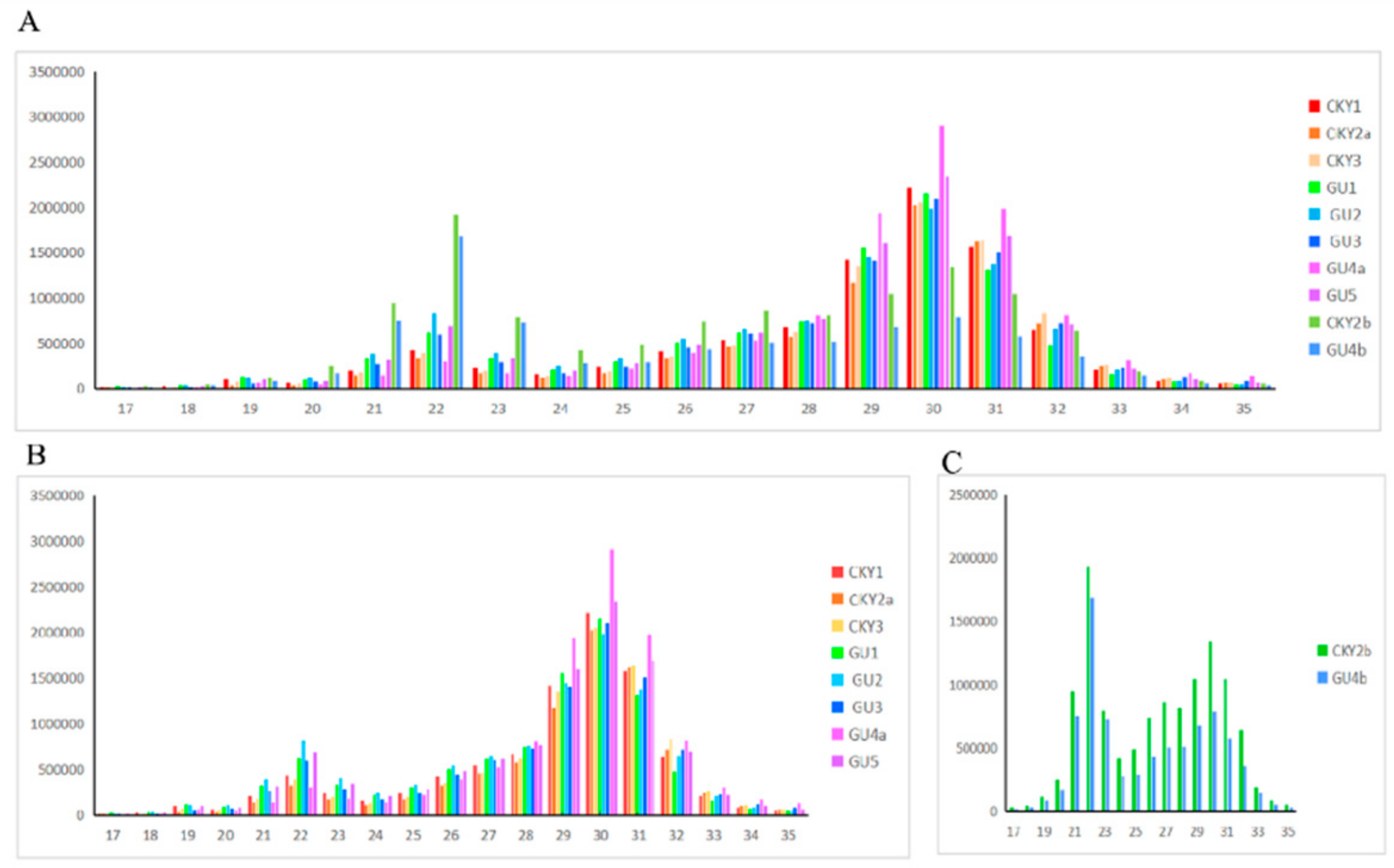
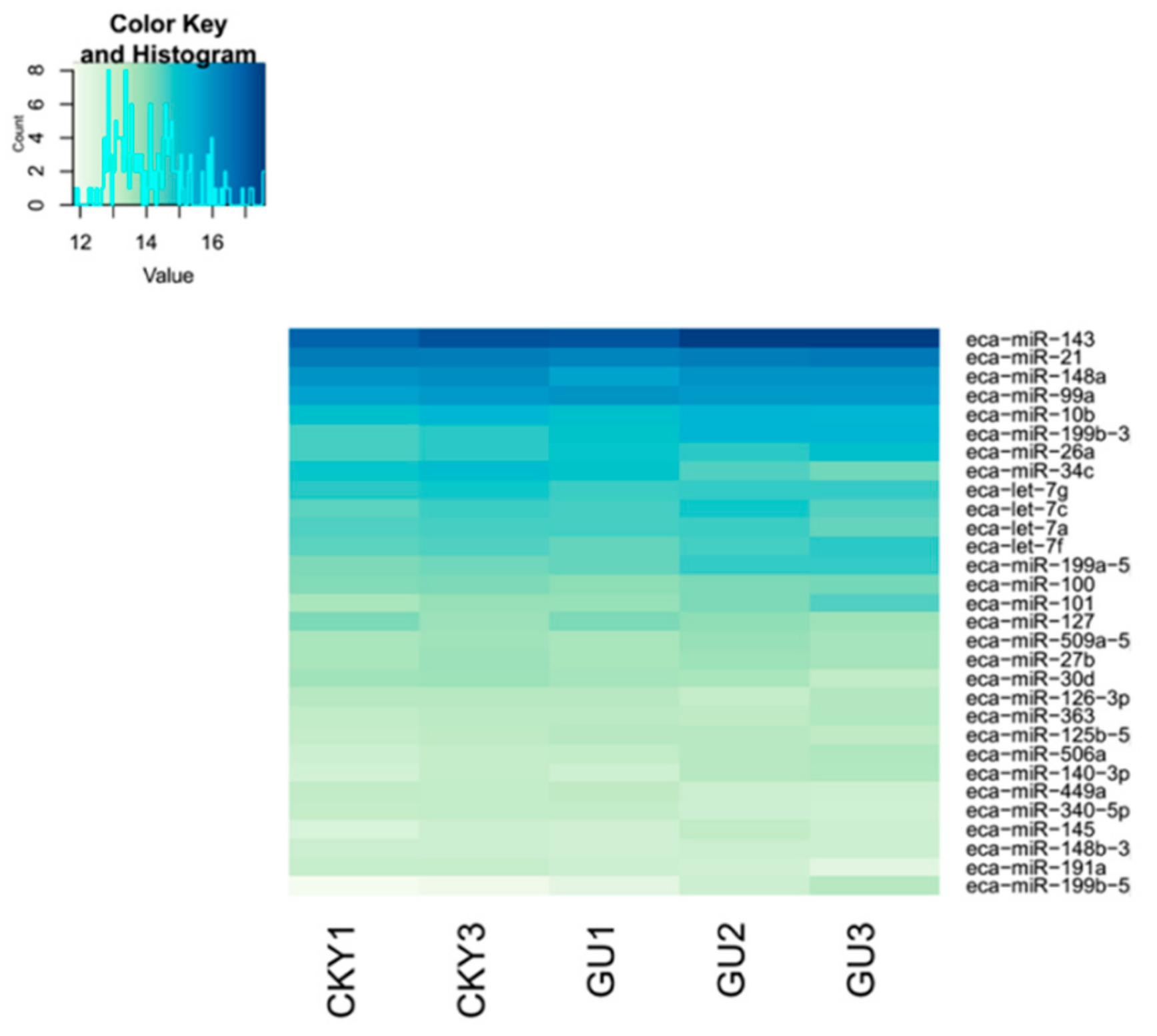
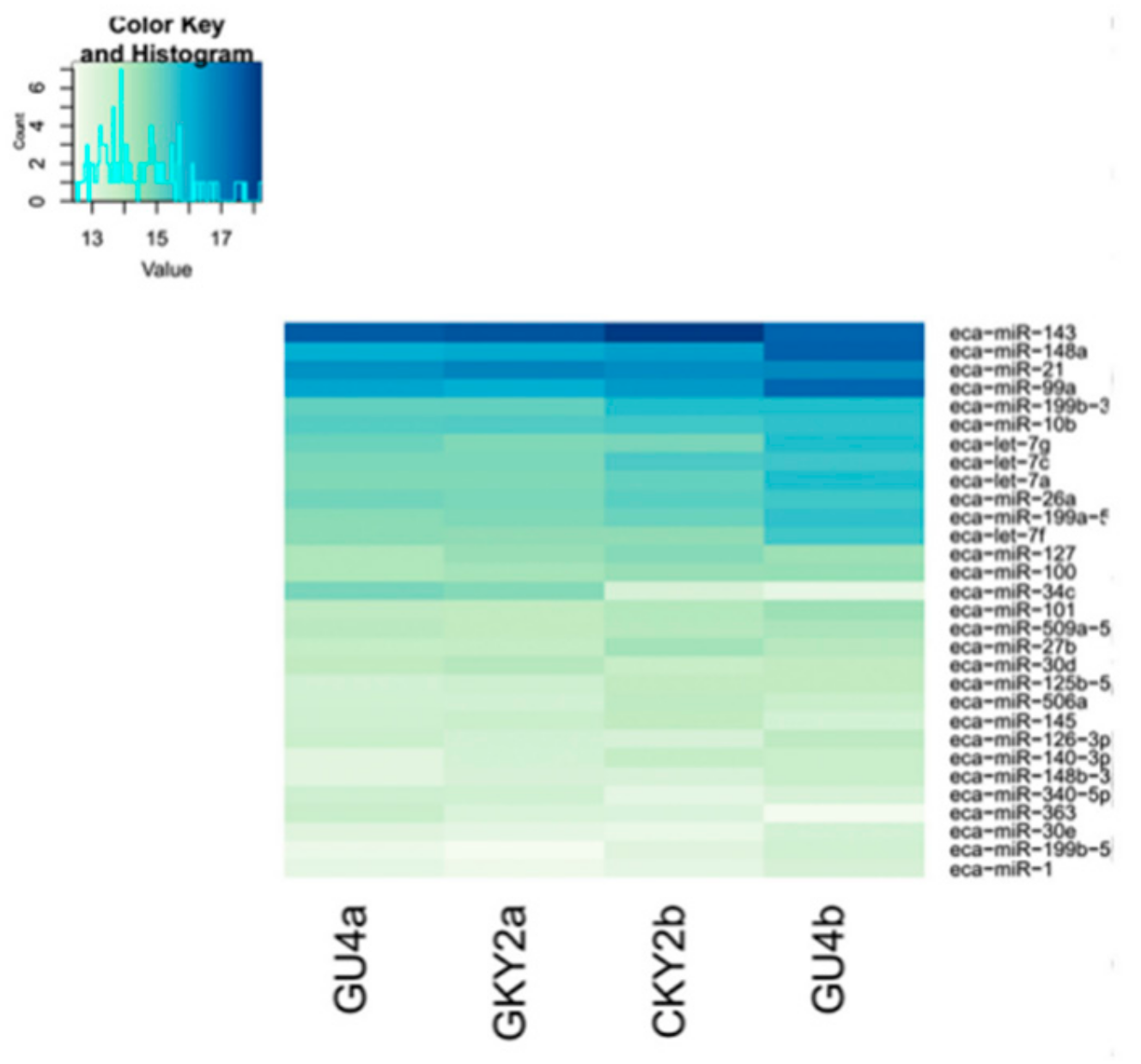
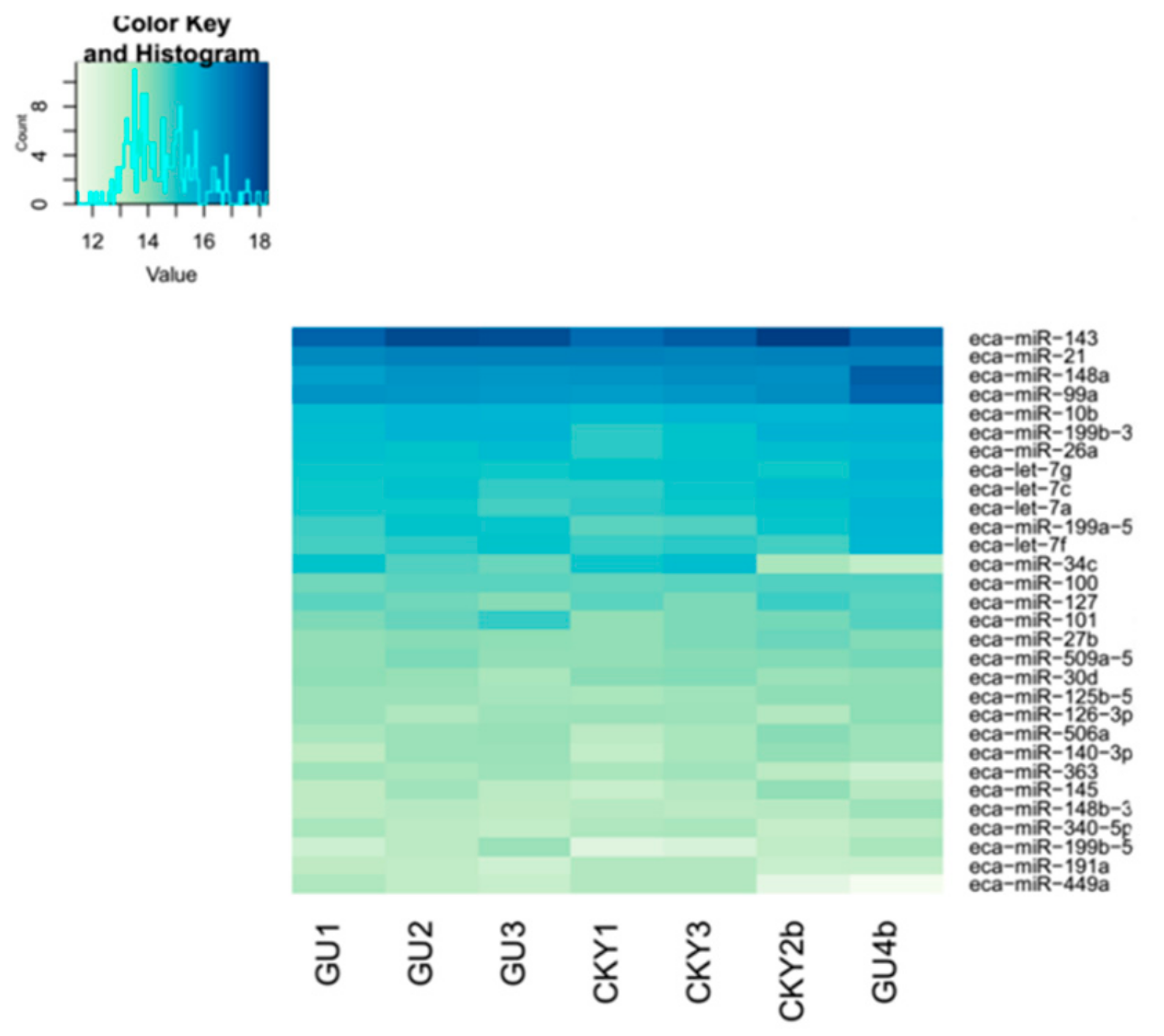
3.1. Construction of the miRNA Expression Profile
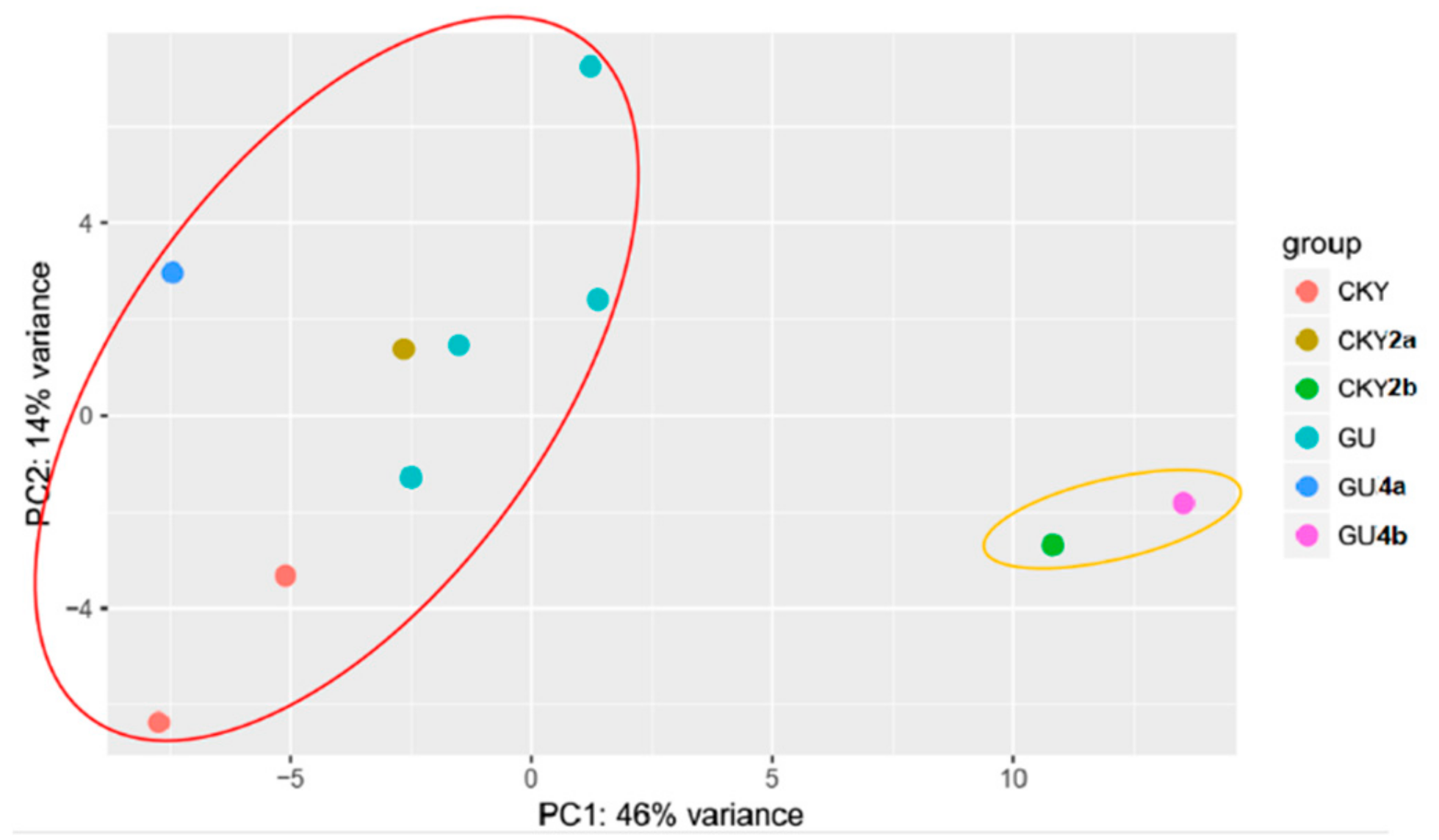
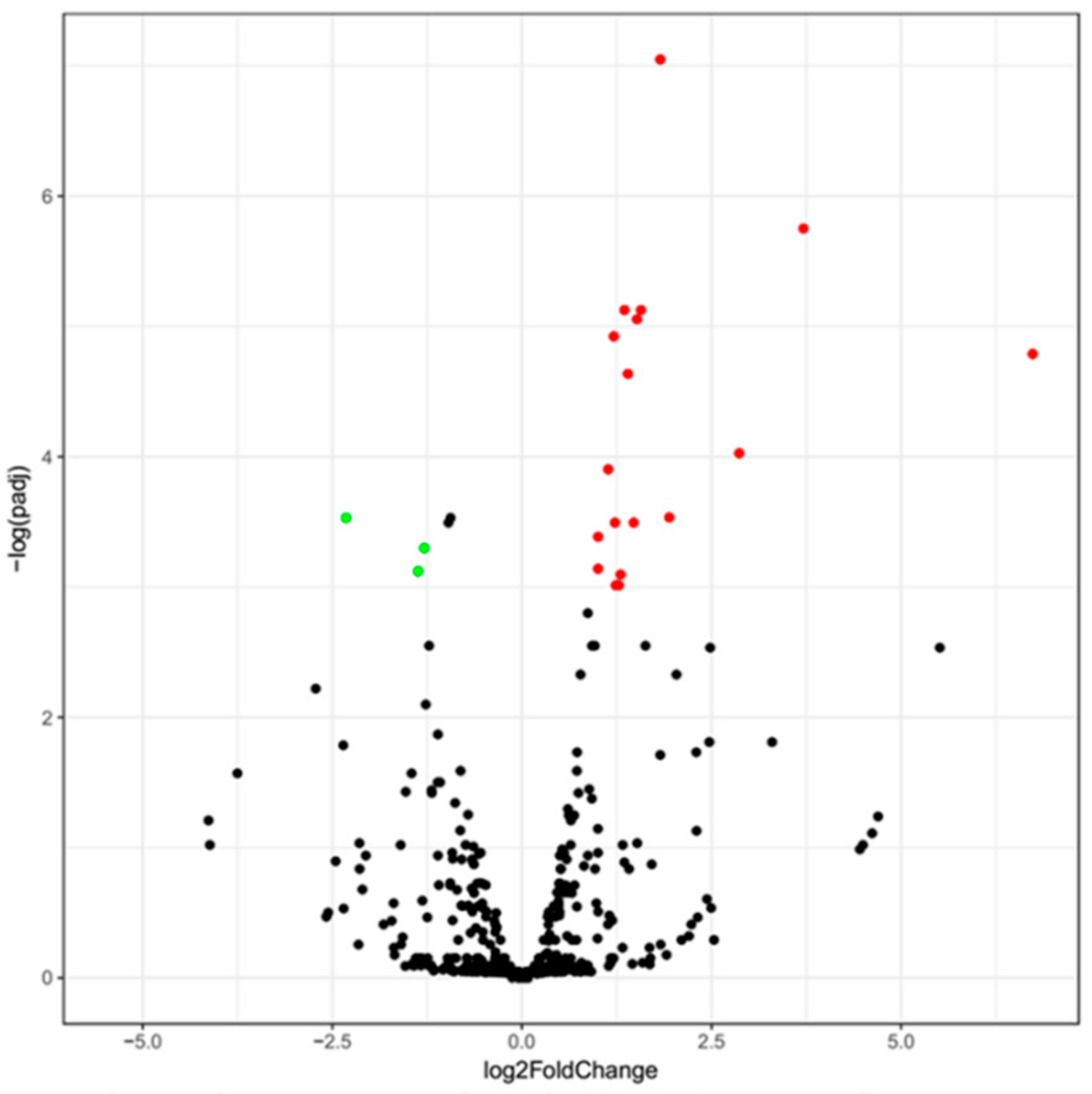
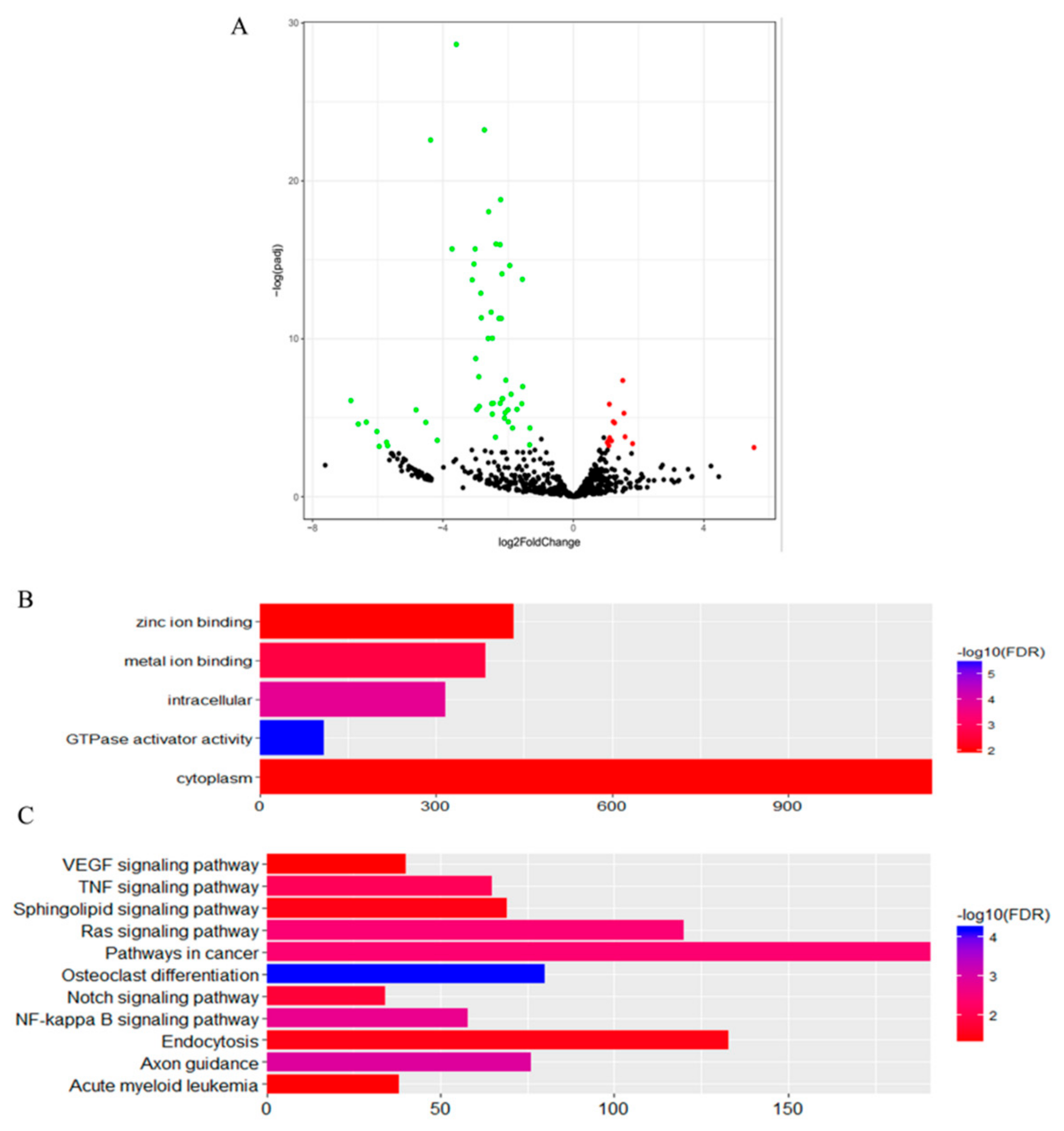
3.2. Differentially Expressed miRNAs of Guanzhong and Chakouyi Horses
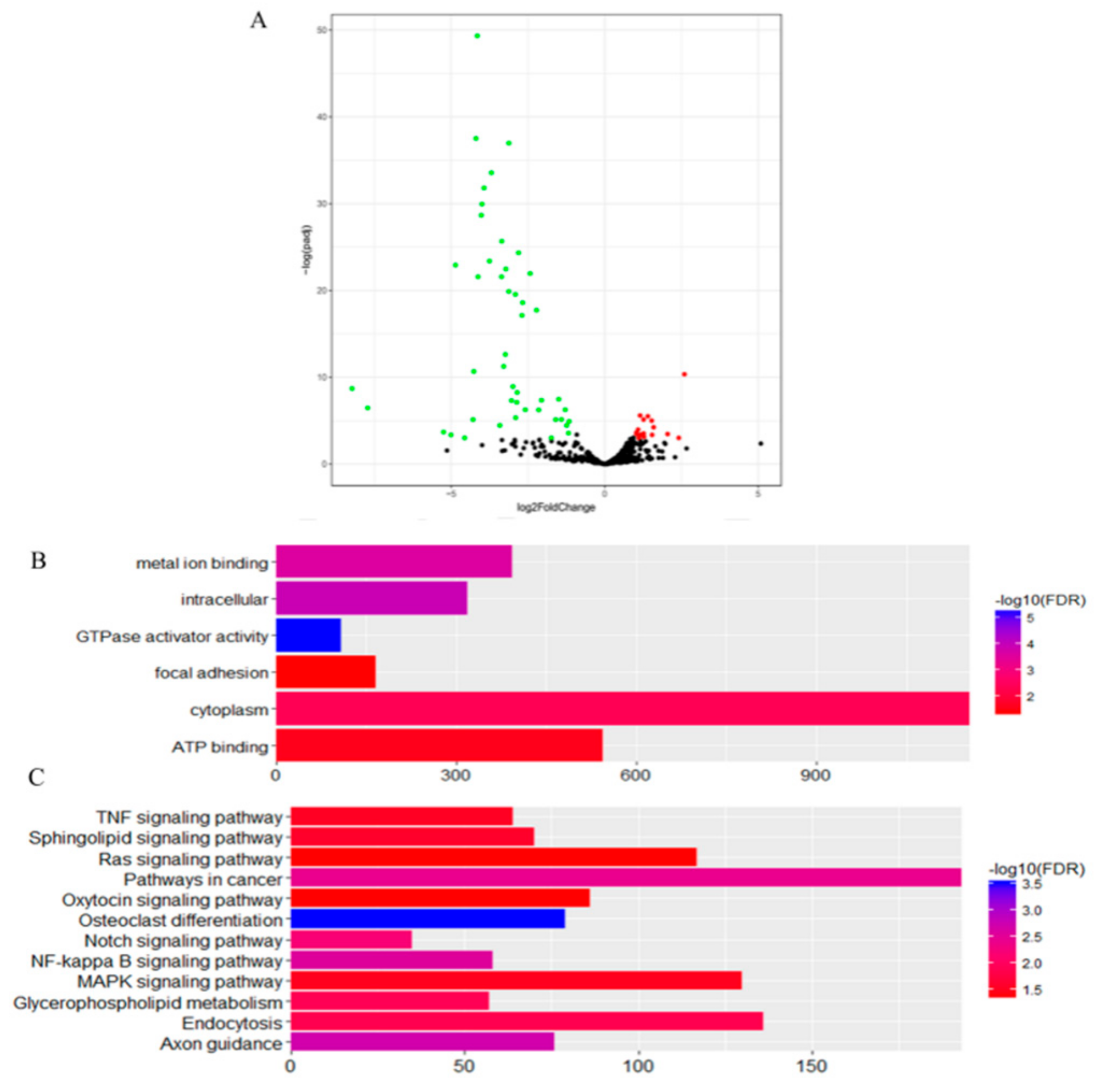
3.3. Differentially Expressed miRNAs of DTs and UDTs
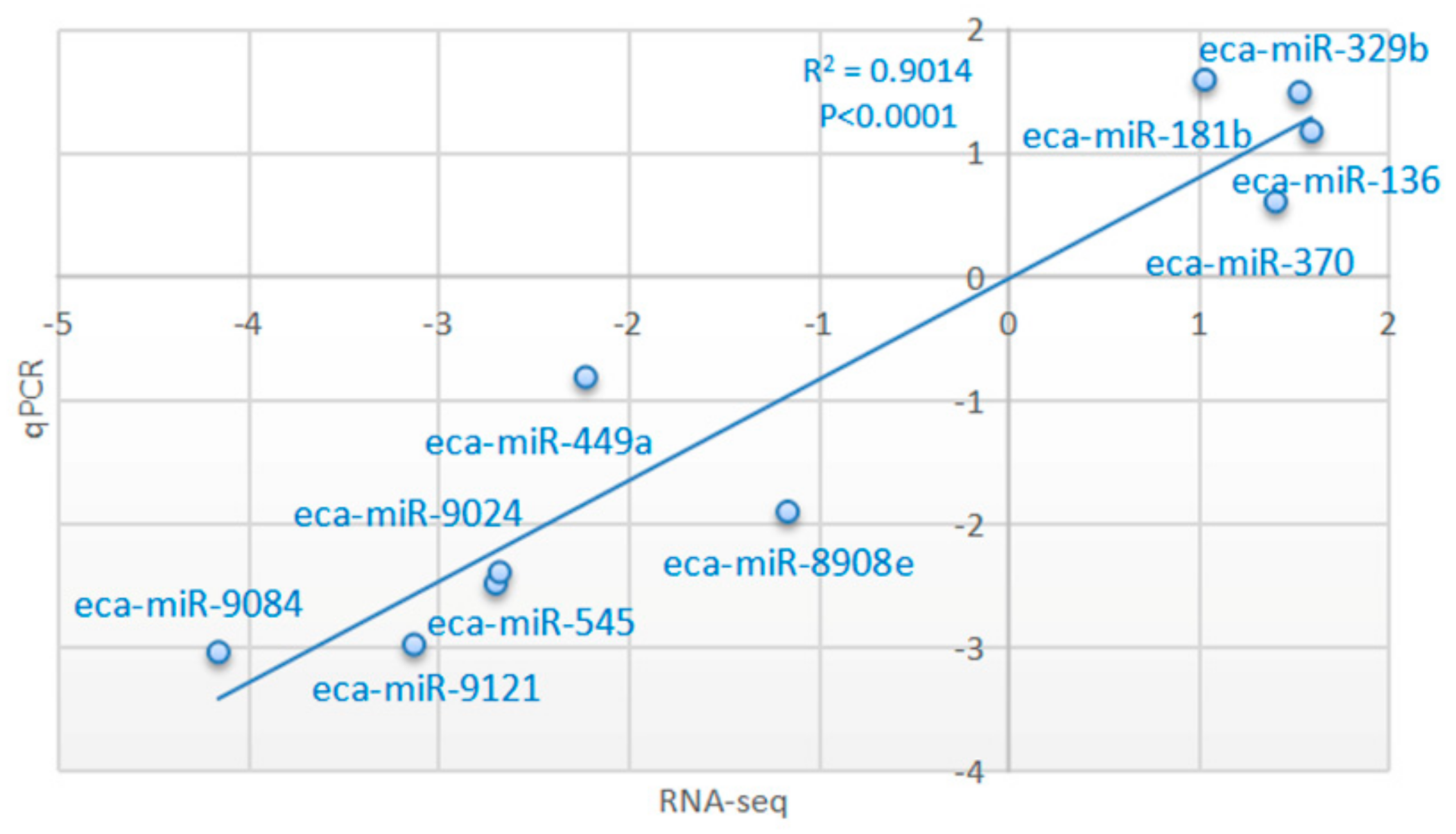
3.4. Validation of Transcriptome Results by qRT-PCR
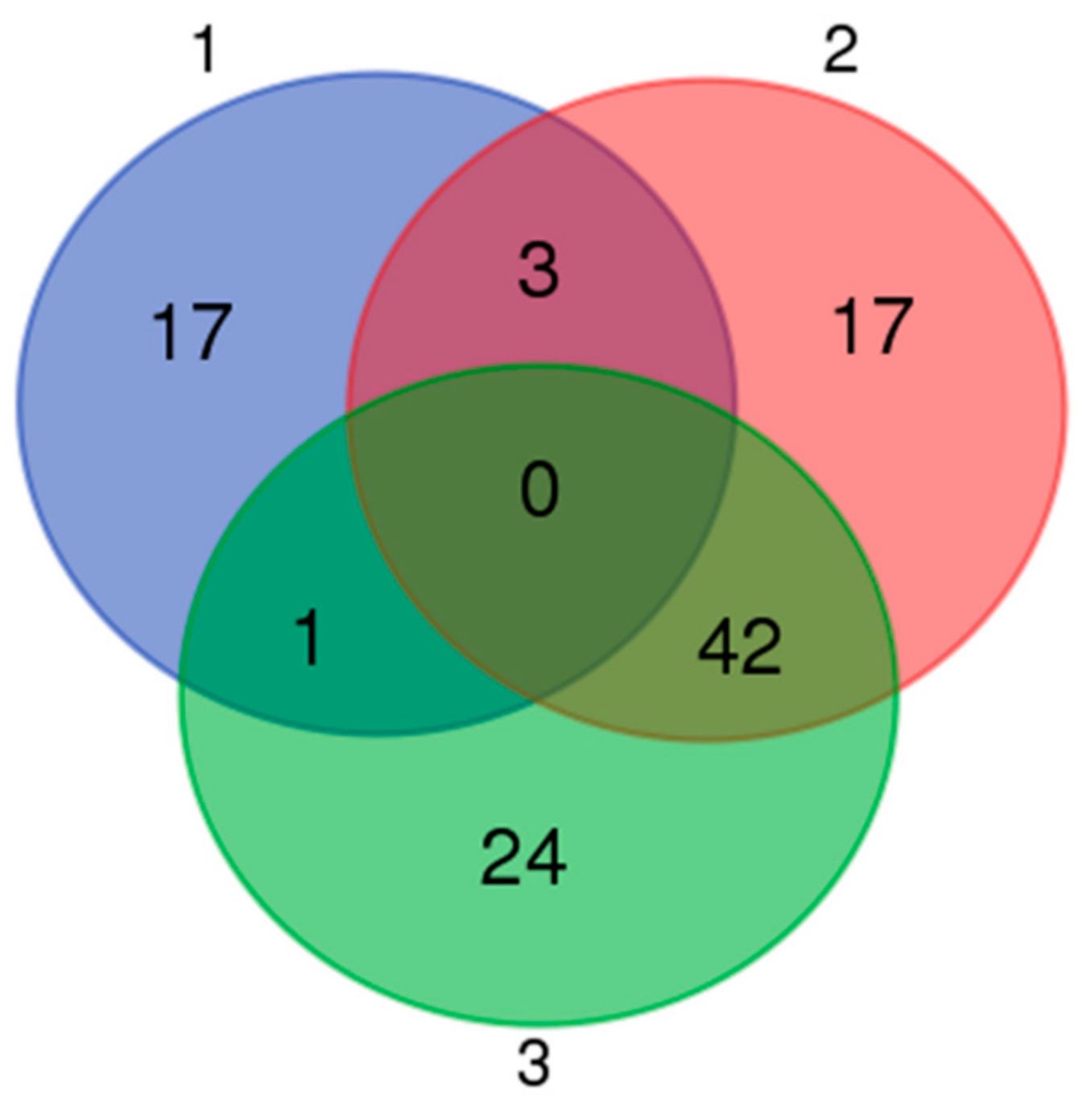
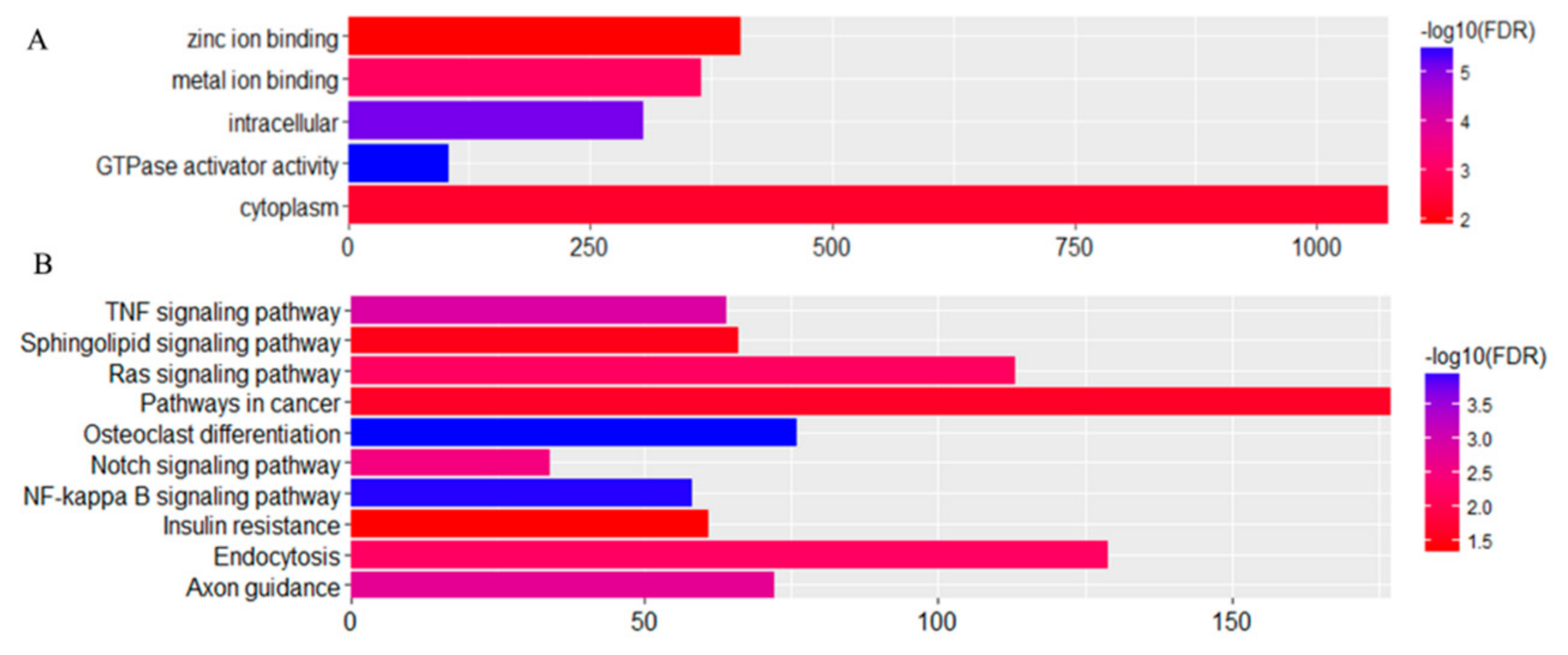
3.5. Expression Profiles of Commonly Differentially Expressed miRNAs in UDTs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability
Animal Ethics
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.; Cheng, H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 2012, 13, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Meyer, J.; Borkhardt, A.; Tuschl, T. New microRNAs from mouse and human. RNA 2003, 9, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of Novel Genes Coding for Small Expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, R.I.; Yan, K.P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Kloosterman, W.P.; Plasterk, R.H. The diverse functions of microRNAs in animal development and disease. Dev. Cell 2006, 11, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Stefani, G.; Slack, F.J. Small non-coding RNAs in animal development. Nat. Rev. Mol. Cell Biol. 2008, 9, 219–230. [Google Scholar] [CrossRef]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef] [Green Version]
- Maatouk, D.M.; Loveland, K.L.; McManus, M.T. Dicer1 is required for differentiation of the mouse male germline. Biol. Reprod. 2008, 79, 696–703. [Google Scholar] [CrossRef] [Green Version]
- Huff, D.S.; Fenig, D.M.; Canning, D.A.; Carr, M.G.; Zderic, S.A.; Snyder, H.M. Abnormal germ cell development in cryptorchidism. Horm. Res. 2001, 55, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Delaney, D.P.; Kolon, T.F. Gene expression alterations in cryptorchid males using spermatozoal microarray analysis. Fertil. Steril. 2009, 92, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Hadziselimovic, F.; Herzog, B. The importance of both an early orchidopexy and germ cell maturation for fertility. Lancet 2001, 358, 1156–1157. [Google Scholar] [CrossRef]
- Carreau, S.; Lambard, S.; Said, L.; Saad, A.; Galeraud-Denis, I. RNA dynamics of fertile and infertile spermatozoa. Biochem. Soc. Trans. 2007, 35, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Platts, A.E.; Dix, D.J.; Chemes, H.E.; Thompson, K.E.; Goodrich, R.; Rockett, J.C.; Rawe, V.Y.; Quintana, S.; Diamond, M.P.; Strader, L.F.; et al. Success and failure in human spermatogenesis as revealed by teratozoospermic RNAs. Hum. Mol. Genet. 2007, 16, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Garrido, N.; Martínez-Conejero, J.A.; Jauregui, J.; Horcajadas, J.A.; Simón, C.; Remohí, J.; Meseguer, M. Microarray analysis in sperm from fertile and infertile men without basic sperm analysis abnormalities reveals a significantly different transcriptome. Fertil. Steril. 2009, 91, 1307–1310. [Google Scholar] [CrossRef]
- Bissonnette, N.; Levesque-Sergerie, J.P.; Thibault, C.; Boissonneault, G. Spermatozoal transcriptome profiling for bull sperm motility: A potential tool to evaluate semen quality. Reproduction 2009, 138, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.H.; Mitchell, D.A.; McGowan, S.D.; Evanoff, R.; Griswold, M.D. Two miRNA clusters, Mir-17-92 (Mirc1) and Mir-106b-25(Mirc3), are involved in the regulation of spermatogonial differentiation in mice. Biol. Reprod. 2012, 86, 72. [Google Scholar] [CrossRef]
- Yang, C.C.; Lin, Y.S.; Hsu, C.C.; Wu, S.C.; Lin, E.C.; Cheng, W.T. Identification and sequencing of remnant messenger RNAs found in domestic swine (Sus scrofa) fresh ejaculated spermatozoa. Anim. Reprod. Sci. 2009, 113, 143–155. [Google Scholar] [CrossRef]
- Yu, Z.; Raabe, T.; Hecht, N.B. MicroRNA Mirn122a reduces expression of the posttranscriptionally regulated germ cell transition protein 2 (Tnp2) messenger RNA (mRNA) by mRNA cleavage. Biol. Reprod. 2005, 73, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Moritoki, Y.; Hayashi, Y.; Mizuno, K.; Kamisawa, H.; Nishio, H.; Kurokawa, S.; Ugawa, S.; Kojima, Y.; Kohri, K. Expression profiling of microRNA in cryptorchid testes: miR-135a contributes to the maintenance of spermatogonial stem cells by regulating FoxO1. J. Urol. 2014, 191, 1174–1180. [Google Scholar] [CrossRef]
- Das, P.J.; Fiona, M.C.; Monika, V.; Nandina, P.; Cathy, G.; Gang, L.; Priyanka, K.; Sudderth, A.K.; Teague, S.; Love, C.C.; et al. Stallion Sperm Transcriptome Comprises Functionally Coherent Coding and Regulatory RNAs as Revealed by Microarray Analysis and RNA-seq. PLoS ONE 2013, 8, e56535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Urh, K.; Kunej, T. Molecular mechanisms of cryptorchidism development: Update of the database, disease comorbidity, and initiative for standardization of reporting in scientific literature. Andrology 2016, 4, 894–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paria, N.; Raudsepp, T.; Pearks Wilkerson, A.J.; O’Brien, P.C.; Ferguson-Smith, M.A.; Love, C.C.; Arnold, C.; Rakestraw, P.; Murphy, W.J.; Chowdhary, B.P. A gene catalogue of the euchromatic male-specific region of the horse Y chromosome: Comparison with human and other mammals. PLoS ONE 2011, 6, e21374. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Qian, L.; Yang, L. Diverse expression of microRNA precursors in human cancer cell lines. Cancer Res. 2004, 45, 64. [Google Scholar]
- Vasileva, A.; Tiedau, D.; Firooznia, A.; Müller-Reichert, T.; Jessberger, R. Tdrd6 is required for spermiogenesis, chromatoid body architecture, and regulation of miRNA expression. Curr. Biol. 2009, 19, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Buchold, G.M.; Coarfa, C.; Kim, J.; Milosavljevic, A.; Gunaratne, P.H.; Matzuk, M.M. Analysis of microRNA expression in the prepubertal testis. PLoS ONE 2010, 5, e15317. [Google Scholar] [CrossRef] [Green Version]
- Niu, Z.; Goodyear, S.; Rao, S.; Wu, X.; Tobias, J.W.; Avarbock, M.R.; Brinster, R.L. MicroRNA-21 regulates the self-renewal of mouse spermatogonial stem cells. PNAS 2011, 108, 12740–12745. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Tsai-Morris, C.H.; Sato, H.; Villar, J.; Kang, J.H.; Zhang, J.M.L. Testis-specific miRNA-469 up-regulated in gonad otropinregulated testicular RNA helicase (GRTH/DDX25)-null mice silences transition protein 2 and protamine 2 messages at sites within coding region. Implications of its role in germ cell development. J. Biol. Chem. 2011, 286, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mclver, S.C.; Stanger, S.J.; Santarelli, D.M.; Roman, S.D.; Nixon, B.; McLaughlin, E.A. A unique combination of male germ cell miRNAs coordinates gonocyte differentiation. PLoS ONE 2012, 7, e35553. [Google Scholar]
- Hayashi, T.; Kageyama, Y.; Ishizaka, K.; Xia, G.; Kihara, K.; Oshima, H. Requirement of Notch 1 and its ligand jagged 2 expressions for spermatogenesis in rat and human testes. J. Androl. 2001, 22, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.M.; Pang, R.T.K.; Chiu, P.C.N.; Wong, B.P.C.; Lao, K.; Lee, K.F.; Yeung, W.S.B. Sperm-borne microRNA-34c is required for the first cleavage division in mouse. PNAS 2012, 106, 490–494. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, C.M.; Hikim, A.P.; Lue, Y.; Portugal, A.M.; Guo, T.B.; Hsu, S.Y.; Salameh, W.A.; Wang, C.; Hsueh, A.J.; Swerdloff, R.S. Impairment of spermatogenesis in transgenic mice with selective overexpression of Bcl-2 in the somatic cells of the testis. J. Androl. 2001, 22, 981–991. [Google Scholar] [CrossRef]
- Selbach, M.; Schwanhäusser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 7209, 58–63. [Google Scholar] [CrossRef]
- Yan, N.; Lu, Y.; Sun, H.; Qiu, W.; Tao, D.; Liu, Y.; Chen, H.; Yang, Y.; Zhang, S.; Li, X.; et al. Microarray profiling of microRNAs expressed in testis tissues of developing primates. J. Assist. Reprod. Genet. 2009, 26, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Zhang, Y.L.; Buchold, G.M.; Jetten, A.M.; O’Brien, D.A. Analysis of germ cell nuclear factor transcripts and protein expression during spermatogenesis. Biol. Reprod. 2003, 68, 1620–1630. [Google Scholar] [CrossRef]
- Connor, F.; Wright, E.; Denny, P.; Koopman, P.; Ashworth, A. The Sryrelated HMG box-containing gene Sox6 is expressed in the adult testis and developing nervous system of the mouse. Nucleic Acids Res. 1995, 23, 3365–3372. [Google Scholar] [CrossRef] [Green Version]
- Serge, C.; Rex, A.H. Oestrogens and spermatogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1517–1535. [Google Scholar]
- Hannema, S.E.; Scott, I.S.; Rajpert De Meyts, E.; Skakkebaek, N.E.; Coleman, N.; Hughes, I.A. Testicular development in the complete androgen insensitivity syndrome. J. Pathol. 2010, 208, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Vandenput, L.; Ederveen, A.; Erben, R.; Stahr, K.; Swinnen, J.V.; Herch, E.V.; Verstuyf, A.; Boonen, S.; Bouillon, R.; Vanderschueren, D. Testosterone prevents orchidectomy-induced bone loss in estrogen receptor-alpha knockout mice. Biochem. Biophys. Res. Commun. 2001, 285, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Franzoso, G. Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 1997, 11, 3482–3496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, A.; Sachidanandam, R.; Hannon, G.J.; Carmell, M.A. A germlinespecific class of small RNAs binds mammalian Piwi proteins. Nature 2006, 442, 199–202. [Google Scholar] [CrossRef]
- Kawano, M.; Kawaji, H.; Grandjean, V.; Kiani, J.; Rassoulzadegan, M. Novel small noncoding RNAs in mouse spermatozoa, zygotes and early embryos. PLoS ONE 2012, 7, e44542. [Google Scholar] [CrossRef]
- Sai Lakshmi, S.; Agrawal, S. piRNABank: A web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res. 2008, 36, D173–D177. [Google Scholar] [CrossRef]
- Thomson, T.; Lin, H. The biogenesis and function of PIWI proteins and piRNAs: Progress and prospect. Annu. Rev. Cell Dev. Biol. 2009, 25, 355–376. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Breed | Age | Testis |
|---|---|---|---|
| GU1 | Guanzhong | 2 years | Descended |
| GU2 | Guanzhong | 2 years | Descended |
| GU3 | Guanzhong | 2 years | Descended |
| GU4a | Guanzhong | 2 years | Descended |
| GU4b | Guanzhong | 2 years | Undescended |
| GU5 | Guanzhong | 2 years | Descended |
| CKY1 | Chakouyi | 5 years | Descended |
| CKY2a | Chakouyi | 3 years | Descended |
| CKY2b | Chakouyi | 3 years | Undescended |
| CKY3 | Chakouyi | 4 years | Descended |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, H.; Chen, Q.; Gao, Y.; Li, J.; Li, W.; Dang, R.; Lei, C. Comparative Transcriptomics Analysis of Testicular miRNA from Cryptorchid and Normal Horses. Animals 2020, 10, 338. https://doi.org/10.3390/ani10020338
Han H, Chen Q, Gao Y, Li J, Li W, Dang R, Lei C. Comparative Transcriptomics Analysis of Testicular miRNA from Cryptorchid and Normal Horses. Animals. 2020; 10(2):338. https://doi.org/10.3390/ani10020338
Chicago/Turabian StyleHan, Haoyuan, Qiuming Chen, Yuan Gao, Jun Li, Wantao Li, Ruihua Dang, and Chuzhao Lei. 2020. "Comparative Transcriptomics Analysis of Testicular miRNA from Cryptorchid and Normal Horses" Animals 10, no. 2: 338. https://doi.org/10.3390/ani10020338
APA StyleHan, H., Chen, Q., Gao, Y., Li, J., Li, W., Dang, R., & Lei, C. (2020). Comparative Transcriptomics Analysis of Testicular miRNA from Cryptorchid and Normal Horses. Animals, 10(2), 338. https://doi.org/10.3390/ani10020338