Exploring the Rumen and Cecum Microbial Community from Fetus to Adulthood in Goat

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval and Consent to Participate
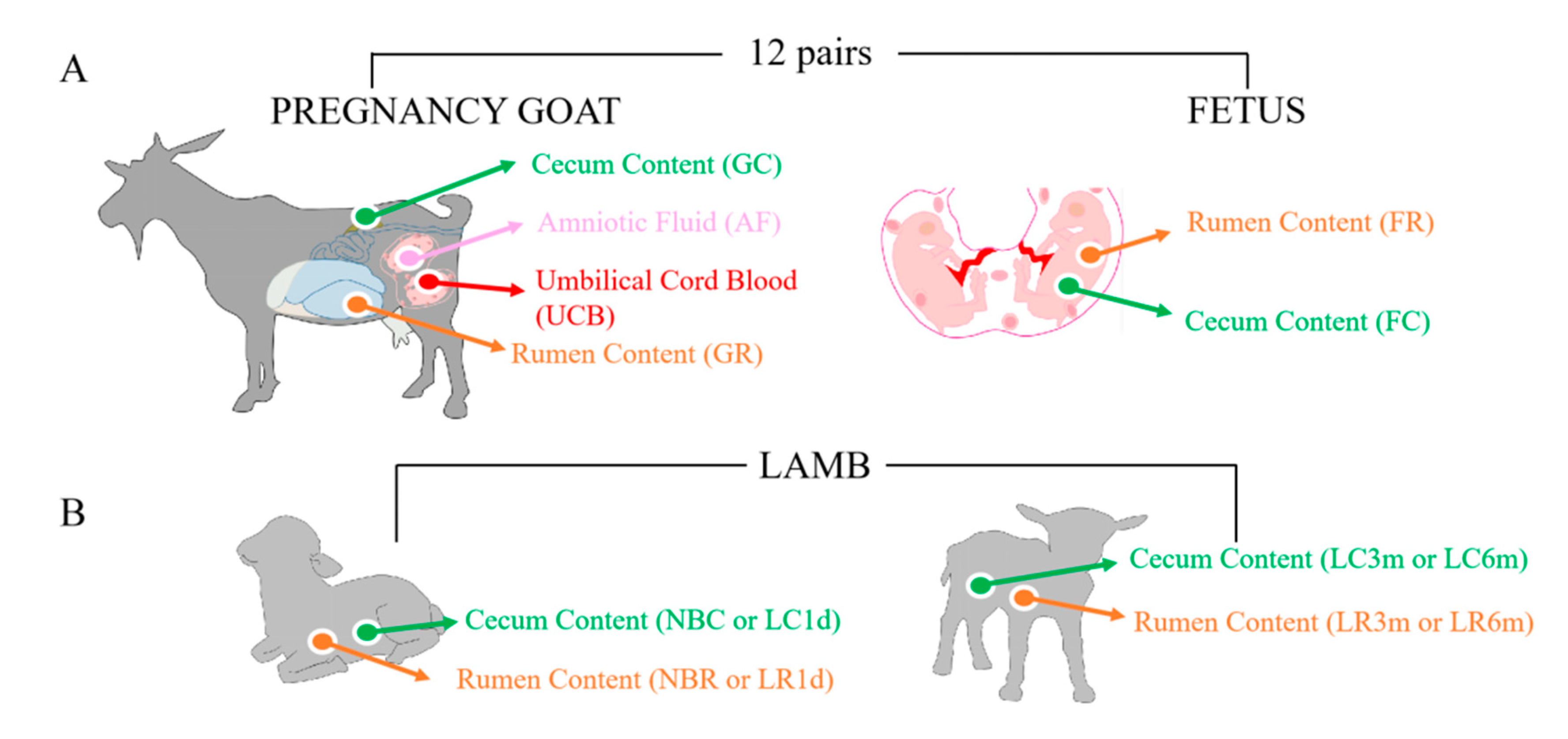
2.2. Study Design and Animal Sampling
2.3. Sequencing and Sequence Analysis
2.4. Taxonomic Classification and Statistical Analyses
2.5. Prediction of Microbial Function
2.6. Statistical Analysis
3. Results
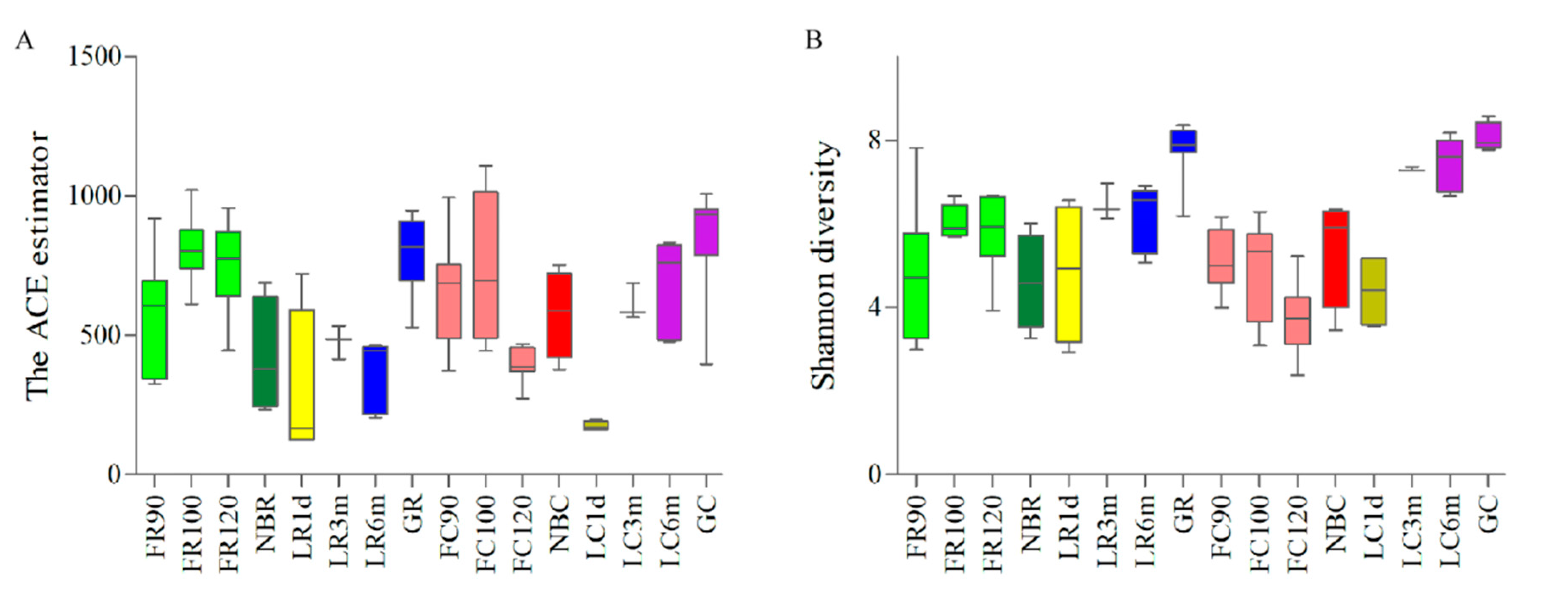
3.1. 16S rRNA Gene Sequencing to Study the Establishment and Dynamic Fluctuations of the Goat Gut Microbiota
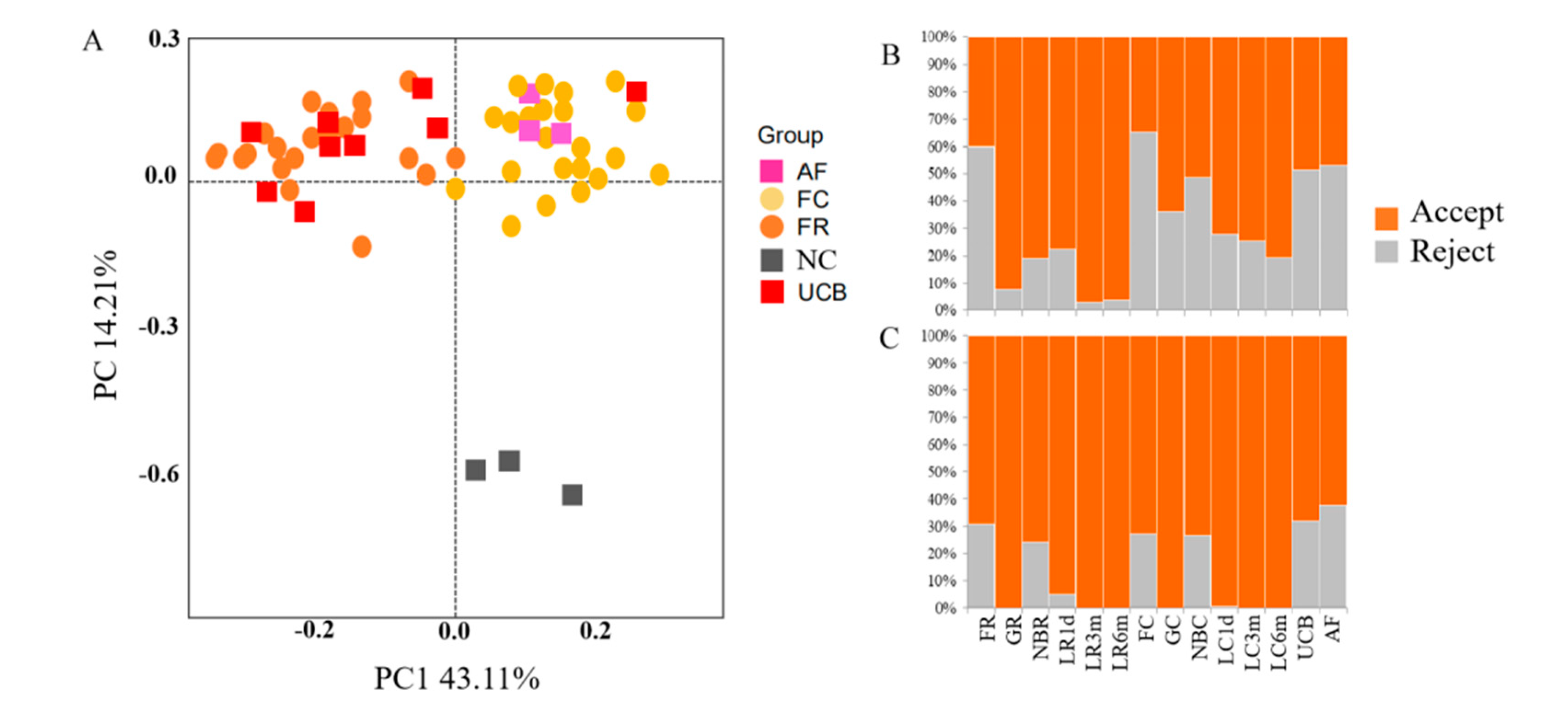
3.2. Contamination Control and Removal of Potential Contaminant Sequences from 16S rDNA Amplicon Sequencing Data
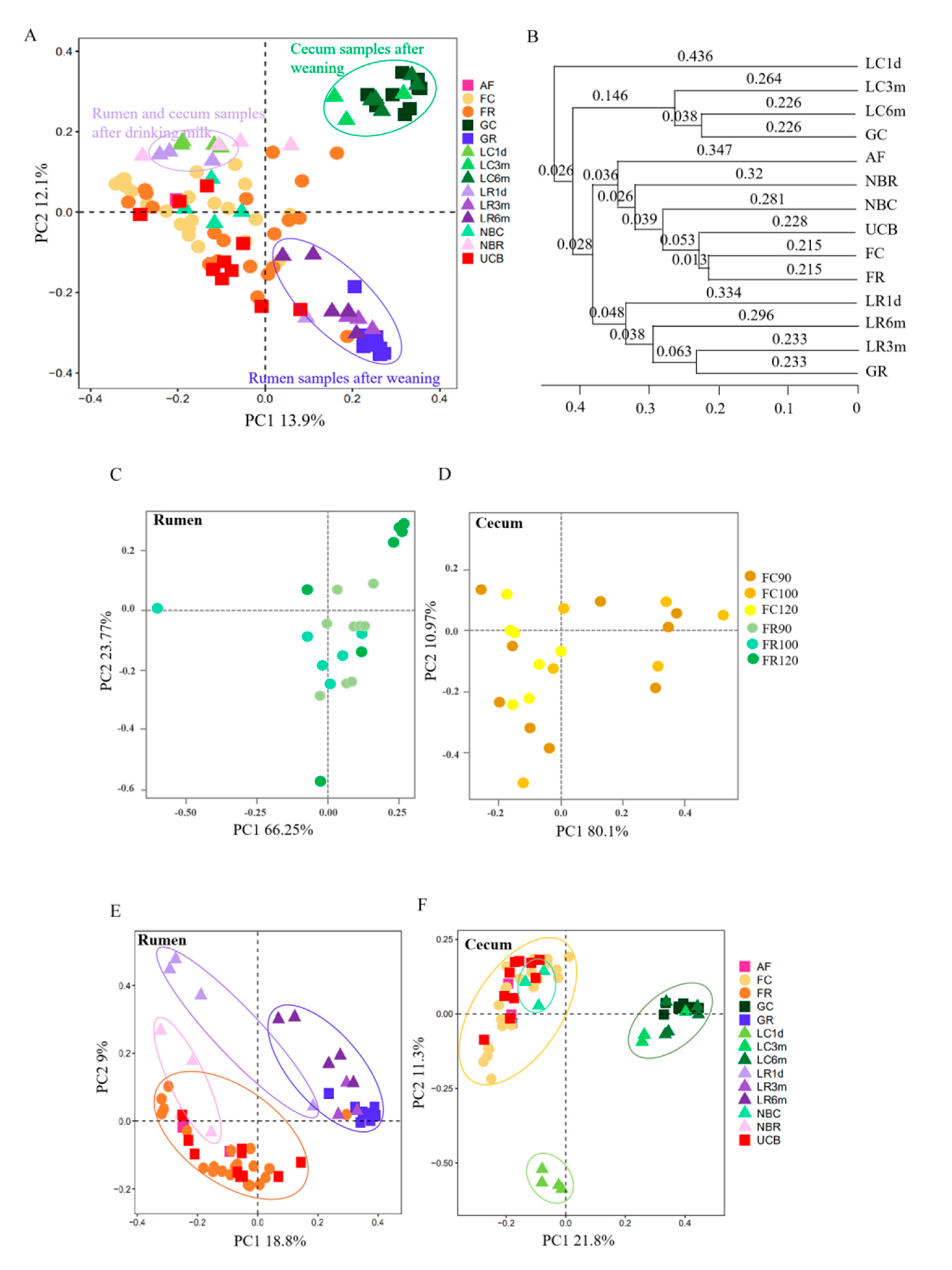
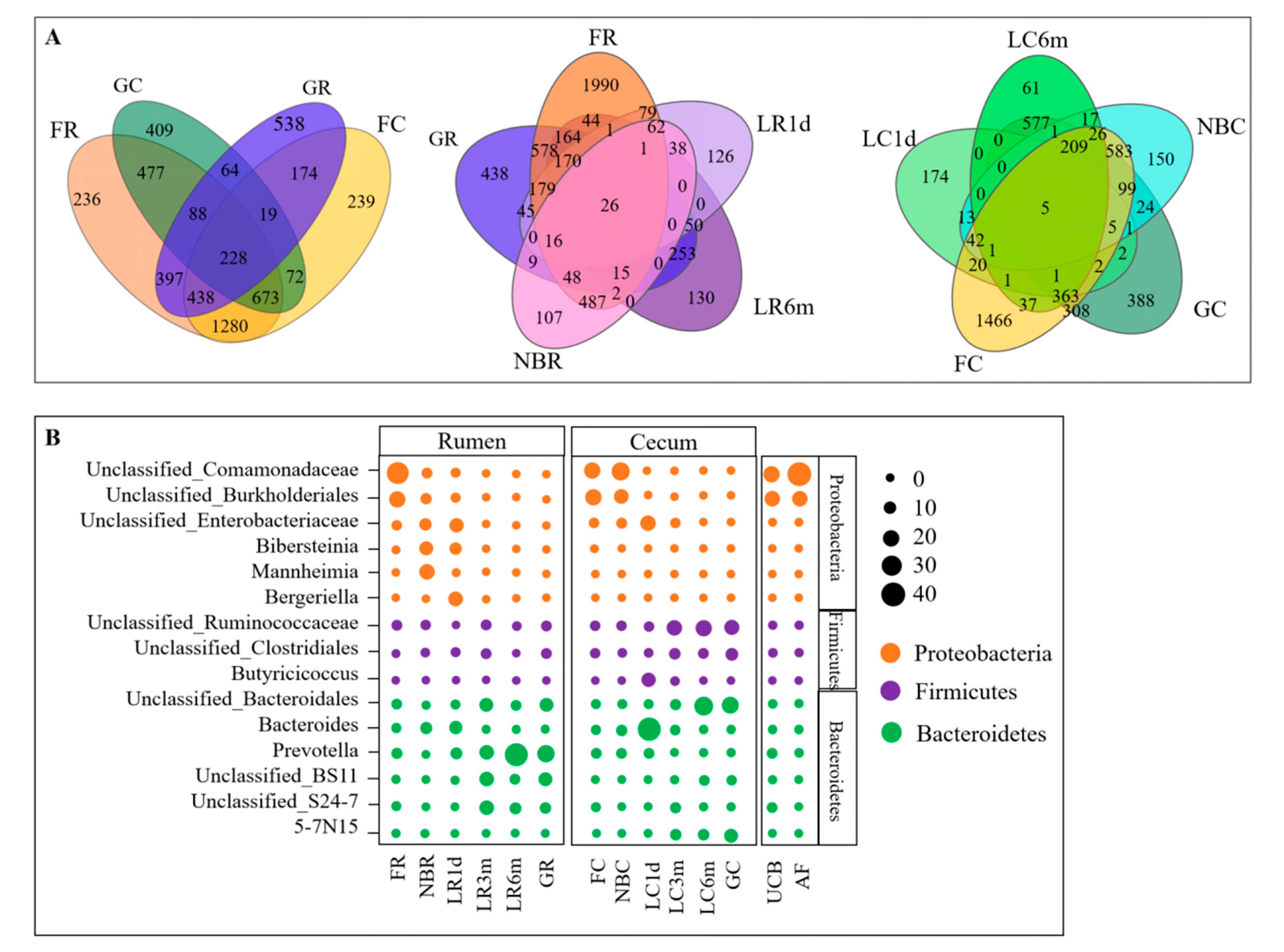
3.3. Microbial Transmission from Fetus to Adulthood
3.4. Rumen and Cecum Microbiomes of Nine Female Goats and Their Fetuses during Late Pregnancy
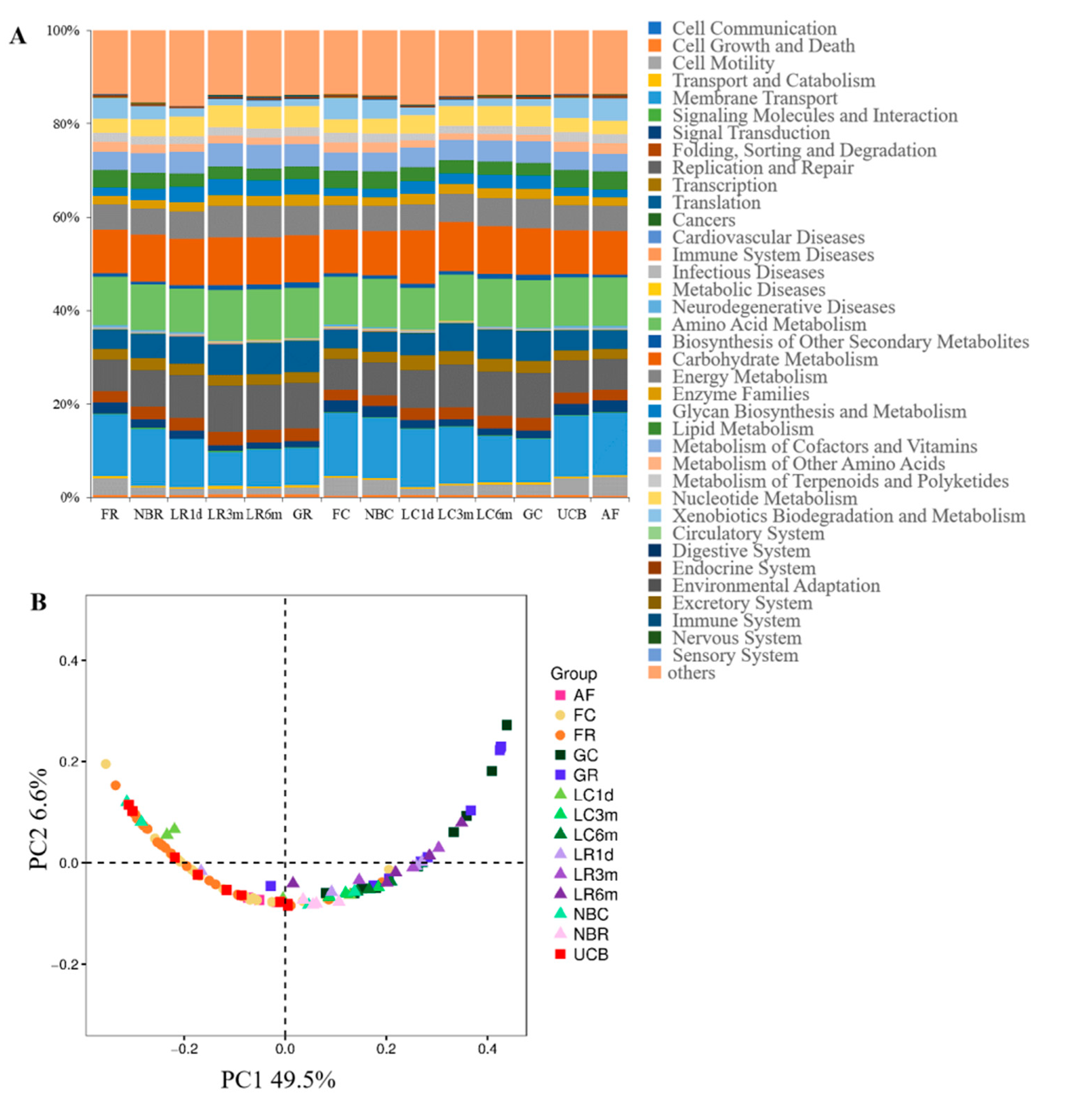
3.5. Predicted Function of Bacteria in Different Developmental Stages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mackie, R.I. Mutualistic fermentative digestion in the gastrointestinal tract: Diversity and evolution. Integr. Comp. Biol. 2002, 42, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.J. Rumen microbiology, biotechnology and ruminant nutrition: The application of research findings to a complex microbial ecosystem. FEMS Microbiol. Lett. 1992, 100, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Bretschger, O.; Osterstock, J.B.; Pinchak, W.E.; Ishii, S.; Nelson, K.E. Microbial fuel cells and microbial ecology: Applications in ruminant health and production research. Microb. Ecol. 2010, 59, 415–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beharka, A.A.; Nagaraja, T.G.; Morrill, J.L.; Kennedy, G.A.; Klemm, R.D. Effects of form of the diet on anatomical, microbial, and fermentative development of the rumen of neonatal calves. J. Dairy Sci. 1998, 81, 1946–1955. [Google Scholar] [CrossRef]
- Li, S.; Khafipour, E.; Krause, D.O.; Kroeker, A.; Rodriguez-Lecompte, J.C.; Gozho, G.N.; Plaizier, J.C. Effects of subacute ruminal acidosis challenges on fermentation and endotoxins in the rumen and hindgut of dairy cows. J. Dairy Sci. 2012, 95, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, J.; Huang, J.; Zhou, C.; Tan, Z. Taxonomic identification of ruminal epithelial bacterial diversity during rumen development in goats. Appl. Environ. Microbiol. 2015, 81, 3502–3509. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Li, B.; Guo, M.; Liu, G.; Yang, Y.; Wang, X.; Chen, Y.; Zhang, E. Maturation of the goat rumen microbiota involves three stages of microbial colonization. Animals 2019, 9, 1028. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, K.; Li, C.; Wang, X.; Chen, Y.; Yang, Y. Characterization and comparison of microbiota in the gastrointestinal tracts of the goat (Capra hircus) during preweaning development. Front. Microbiol. 2019, 10, 2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, J.; Li, X.; Beauchemin, K.A.; Tan, Z.; Tang, S.; Zhou, C. Rumen development process in goats as affected by supplemental feeding v. grazing: Age-related anatomic development, functional achievement and microbial colonisation. Br. J. Nutr. 2015, 113, 888–900. [Google Scholar] [CrossRef] [Green Version]
- Borghi, E.; Massa, V.; Severgnini, M.; Fazio, G.; Avagliano, L.; Menegola, E.; Bulfamante, G.P.; Morace, G.; Borgo, F. Antenatal microbial colonization of mammalian gut. Reprod. Sci. 2019, 26, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Dietz, M.W.; Salles, J.F.; Hsu, B.Y.; Dijkstra, C.; Groothuis, T.; van der Velde, M.; Verkuil, Y.I.; Tieleman, B.I. Prenatal transfer of gut bacteria in rock pigeon. Microorganisms 2019, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Dai, R.; Yang, L.; He, C.; Xu, K.; Liu, S.; Zhao, W.; Xiao, L.; Luo, L.; Zhang, Y.; et al. Inheritance and establishment of gut microbiota in chickens. Front. Microbiol. 2017, 8, 1967. [Google Scholar] [CrossRef]
- Jimenez, E.; Marin, M.L.; Martin, R.; Odriozola, J.M.; Olivares, M.; Xaus, J.; Fernandez, L.; Rodriguez, J.M. Is meconium from healthy newborns actually sterile? Res. Microbiol. 2008, 159, 187–193. [Google Scholar] [CrossRef]
- Yassour, M.; Jason, E.; Hogstrom, L.J.; Arthur, T.D.; Tripathi, S.; Siljander, H.; Selvenius, J.; Oikarinen, S.; Hyoty, H.; Virtanen, S.M.; et al. Strain-level analysis of mother-to-child bacterial transmission during the first few months of life. Cell Host Microbe 2018, 24, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, P.; Pasolli, E.; Tett, A.; Asnicar, F.; Gorfer, V.; Fedi, S.; Armanini, F.; Truong, D.T.; Manara, S.; Zolfo, M.; et al. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 2018, 24, 133–145. [Google Scholar] [CrossRef]
- Mueller, N.T.; Shin, H.; Pizoni, A.; Werlang, I.C.; Matte, U.; Goldani, M.Z.; Goldani, H.; Dominguez-Bello, M.G. Delivery mode and the transition of pioneering gut-microbiota structure, composition and predicted metabolic function. Genes 2017, 8, 364. [Google Scholar] [CrossRef] [Green Version]
- Romano-Keeler, J.; Moore, D.J.; Wang, C.; Brucker, R.M.; Fonnesbeck, C.; Slaughter, J.C.; Li, H.; Curran, D.P.; Meng, S.; Correa, H.; et al. Early life establishment of site-specific microbial communities in the gut. Gut Microbes 2014, 5, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Walker, R.W.; Clemente, J.C.; Peter, I.; Loos, R. The prenatal gut microbiome: Are we colonized with bacteria in utero? Pediatr Obes. 2017, 12, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.; Makino, H.; Cetinyurek, Y.A.; Ben-Amor, K.; Roelofs, M.; Ishikawa, E.; Kubota, H.; Swinkels, S.; Sakai, T.; Oishi, K.; et al. Early-life events, including mode of delivery and type of feeding, siblings and gender, shape the developing gut microbiota. PLoS ONE 2016, 11, e158498. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237r–265r. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, D.M.; Weimer, P.J. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl. Microbiol. Biotechnol. 2007, 75, 165–174. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega, T.R.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Zhong, S.; Ding, Y.; Wang, Y.; Zhou, G.; Guo, H.; Chen, Y.; Yang, Y. Temperature and humidity index (THI)-induced rumen bacterial community changes in goats. Appl. Microbiol. Biotechnol. 2019, 103, 3193–3203. [Google Scholar] [CrossRef]
- Wang, L.; Jin, L.; Xue, B.; Wang, Z.; Peng, Q. Characterizing the bacterial community across the gastrointestinal tract of goats: Composition and potential function. Microbiologyopen 2019, 8, e00820. [Google Scholar] [CrossRef]
- Xue, D.; Chen, H.; Zhao, X.; Xu, S.; Hu, L.; Xu, T.; Jiang, L.; Zhan, W. Rumen prokaryotic communities of ruminants under different feeding paradigms on the Qinghai-Tibetan Plateau. Syst. Appl. Microbiol. 2017, 40, 227–236. [Google Scholar] [CrossRef]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Pitta, D.W.; Pinchak, W.E.; Indugu, N.; Vecchiarelli, B.; Sinha, R.; Fulford, J.D. Metagenomic Analysis of the Rumen Microbiome of Steers with Wheat-Induced Frothy Bloat. Front Microbiol 2016, 7, 689. [Google Scholar] [CrossRef] [Green Version]
- Huo, W.; Zhu, W.; Mao, S. Impact of subacute ruminal acidosis on the diversity of liquid and solid-associated bacteria in the rumen of goats. World J. Microbiol. Biotechnol. 2014, 30, 669–680. [Google Scholar] [CrossRef]
- Rawat, N.; Gilhare, V.R.; Kushwaha, K.K.; Hattimare, D.D.; Khan, F.F.; Shende, R.K.; Jolhe, D.K. Isolation and molecular characterization of Mannheimia haemolytica and Pasteurella multocida associated with pneumonia of goats in Chhattisgarh. Vet. World 2019, 12, 331–336. [Google Scholar] [CrossRef]
- Brogden, K.A.; Lehmkuhl, H.D.; Cutlip, R.C. Pasteurella haemolytica complicated respiratory infections in sheep and goats. Vet. Res. 1998, 29, 233–254. [Google Scholar]
- Devriese, S.; Eeckhaut, V.; Geirnaert, A.; Van den Bossche, L.; Hindryckx, P.; Van de Wiele, T.; Van Immerseel, F.; Ducatelle, R.; De Vos, M.; Laukens, D. Reduced mucosa-associated butyricicoccus activity in patients with ulcerative colitis correlates with aberrant claudin-1 expression. J. Crohns Colitis 2017, 11, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Steppe, M.; Van Nieuwerburgh, F.; Vercauteren, G.; Boyen, F.; Eeckhaut, V.; Deforce, D.; Haesebrouck, F.; Ducatelle, R.; Van Immerseel, F. Safety assessment of the butyrate-producing Butyricicoccus pullicaecorum strain 25-3(T), a potential probiotic for patients with inflammatory bowel disease, based on oral toxicity tests and whole genome sequencing. Food Chem. Toxicol. 2014, 72, 129–137. [Google Scholar] [CrossRef]
- Eeckhaut, V.; Machiels, K.; Perrier, C.; Romero, C.; Maes, S.; Flahou, B.; Steppe, M.; Haesebrouck, F.; Sas, B.; Ducatelle, R.; et al. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 2013, 62, 1745–1752. [Google Scholar] [CrossRef]
- Geirnaert, A.; Steyaert, A.; Van den Abbeele, P.; Eeckhaut, V.; Van Immerseel, F.; Boon, N.; Van de Wiele, T. In vitro characterization of gastrointestinal behavior of Butyricicoccus pullicaecorum, a novel butyrate producing isolate with probiotic potential to counterbalance dysbiosis in inflammatory bowel disease. Commun. Agric Appl. Biol. Sci. 2013, 78, 157–163. [Google Scholar]
- Purushe, J.; Fouts, D.E.; Morrison, M.; White, B.A.; Mackie, R.I.; Coutinho, P.M.; Henrissat, B.; Nelson, K.E. Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: Insights into their environmental niche. Microb. Ecol. 2010, 60, 721–729. [Google Scholar] [CrossRef]
- Belanche, A.; Doreau, M.; Edwards, J.E.; Moorby, J.M.; Pinloche, E.; Newbold, C.J. Shifts in the rumen microbiota due to the type of carbohydrate and level of protein ingested by dairy cattle are associated with changes in rumen fermentation. J. Nutr. 2012, 142, 1684–1692. [Google Scholar] [CrossRef]
- Hook, S.E.; Steele, M.A.; Northwood, K.S.; Dijkstra, J.; France, J.; Wright, A.D.; McBride, B.W. Impact of subacute ruminal acidosis (SARA) adaptation and recovery on the density and diversity of bacteria in the rumen of dairy cows. FEMS Microbiol. Ecol. 2011, 78, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar] [CrossRef] [Green Version]
- Ardissone, A.N.; de la Cruz, D.M.; Davis-Richardson, A.G.; Rechcigl, K.T.; Li, N.; Drew, J.C.; Murgas-Torrazza, R.; Sharma, R.; Hudak, M.L.; Triplett, E.W.; et al. Meconium microbiome analysis identifies bacteria correlated with premature birth. PLoS ONE 2014, 9, e90784. [Google Scholar] [CrossRef] [Green Version]
- Rautava, S.; Collado, M.C.; Salminen, S.; Isolauri, E. Probiotics modulate host-microbe interaction in the placenta and fetal gut: A randomized, double-blind, placebo-controlled trial. Neonatology 2012, 102, 178–184. [Google Scholar] [CrossRef]
- Jimenez, E.; Fernandez, L.; Marin, M.L.; Martin, R.; Odriozola, J.M.; Nueno-Palop, C.; Narbad, A.; Olivares, M.; Xaus, J.; Rodriguez, J.M. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr. Microbiol. 2005, 51, 270–274. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, X.; Liu, G.; Meng, F.; Hong, L.; Li, Y.; Lian, Z.; Yang, Z.; Luo, C.; Liu, D. Exploring the Rumen and Cecum Microbial Community from Fetus to Adulthood in Goat. Animals 2020, 10, 1639. https://doi.org/10.3390/ani10091639
Zou X, Liu G, Meng F, Hong L, Li Y, Lian Z, Yang Z, Luo C, Liu D. Exploring the Rumen and Cecum Microbial Community from Fetus to Adulthood in Goat. Animals. 2020; 10(9):1639. https://doi.org/10.3390/ani10091639
Chicago/Turabian StyleZou, Xian, Guangbin Liu, Fanming Meng, Linjun Hong, Yaokun Li, Zhiquan Lian, Zhenwei Yang, Chenglong Luo, and Dewu Liu. 2020. "Exploring the Rumen and Cecum Microbial Community from Fetus to Adulthood in Goat" Animals 10, no. 9: 1639. https://doi.org/10.3390/ani10091639
APA StyleZou, X., Liu, G., Meng, F., Hong, L., Li, Y., Lian, Z., Yang, Z., Luo, C., & Liu, D. (2020). Exploring the Rumen and Cecum Microbial Community from Fetus to Adulthood in Goat. Animals, 10(9), 1639. https://doi.org/10.3390/ani10091639