Characterization of Bacterial Microbiota Composition along the Gastrointestinal Tract in Rabbits

 ,
,  ,
,  , ,
, ,  , ,
, ,  ,
, 
 , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Microbiota Evaluation
2.2.1. DNA Extraction
2.2.2. 16S Ribosomal RNA (rRNA) Gene Sequencing
2.2.3. Sequence Analysis
2.2.4. Alpha and Beta Diversity Indices
2.3. Statistical Analysis
2.4. Software
3. Results
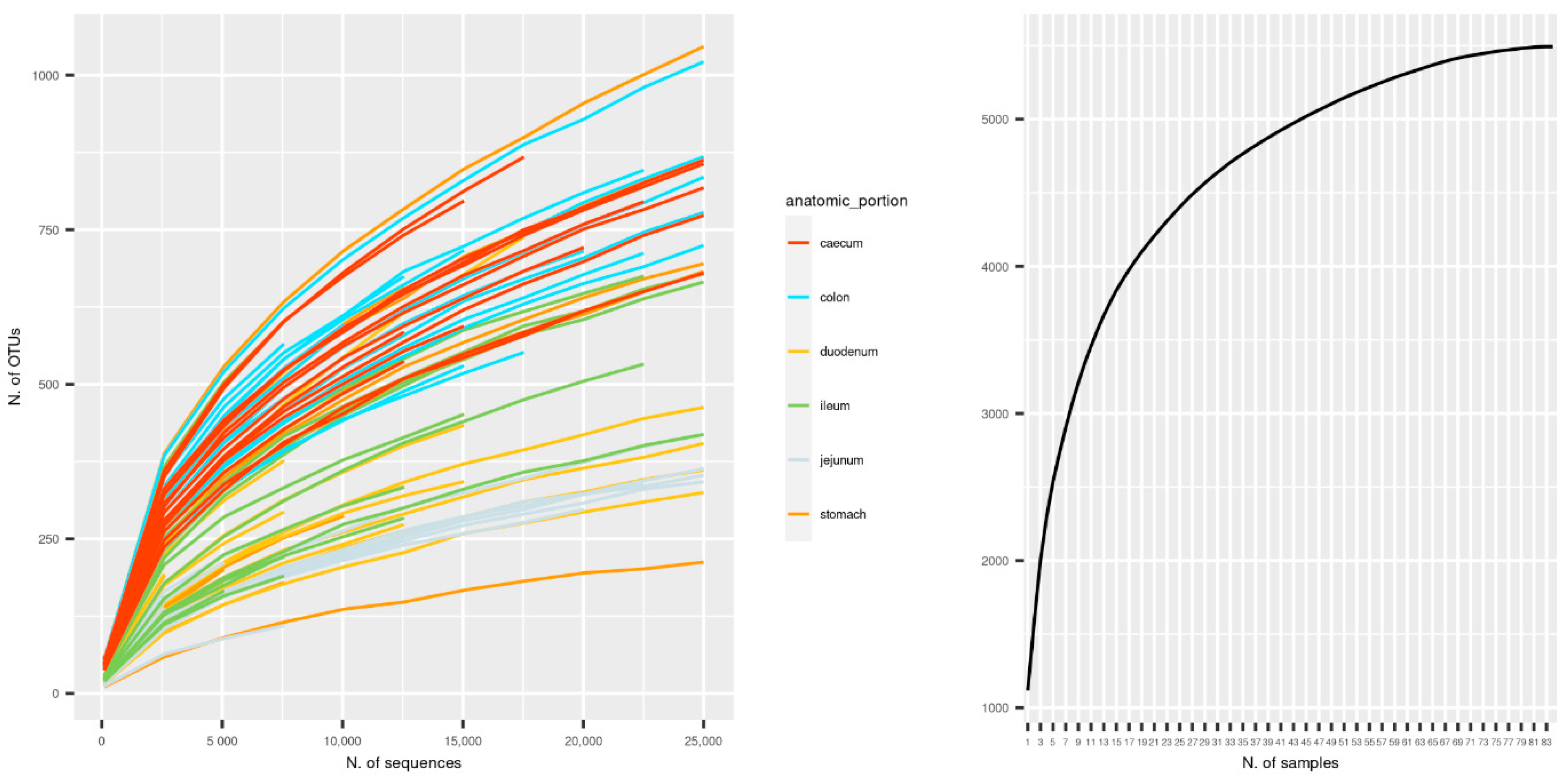
3.1. Sequencing Metrics
3.2. Taxonomic Characterization of Gut Microbiota Composition along the Gastrointestinal Tract
3.3. Core Microbiota
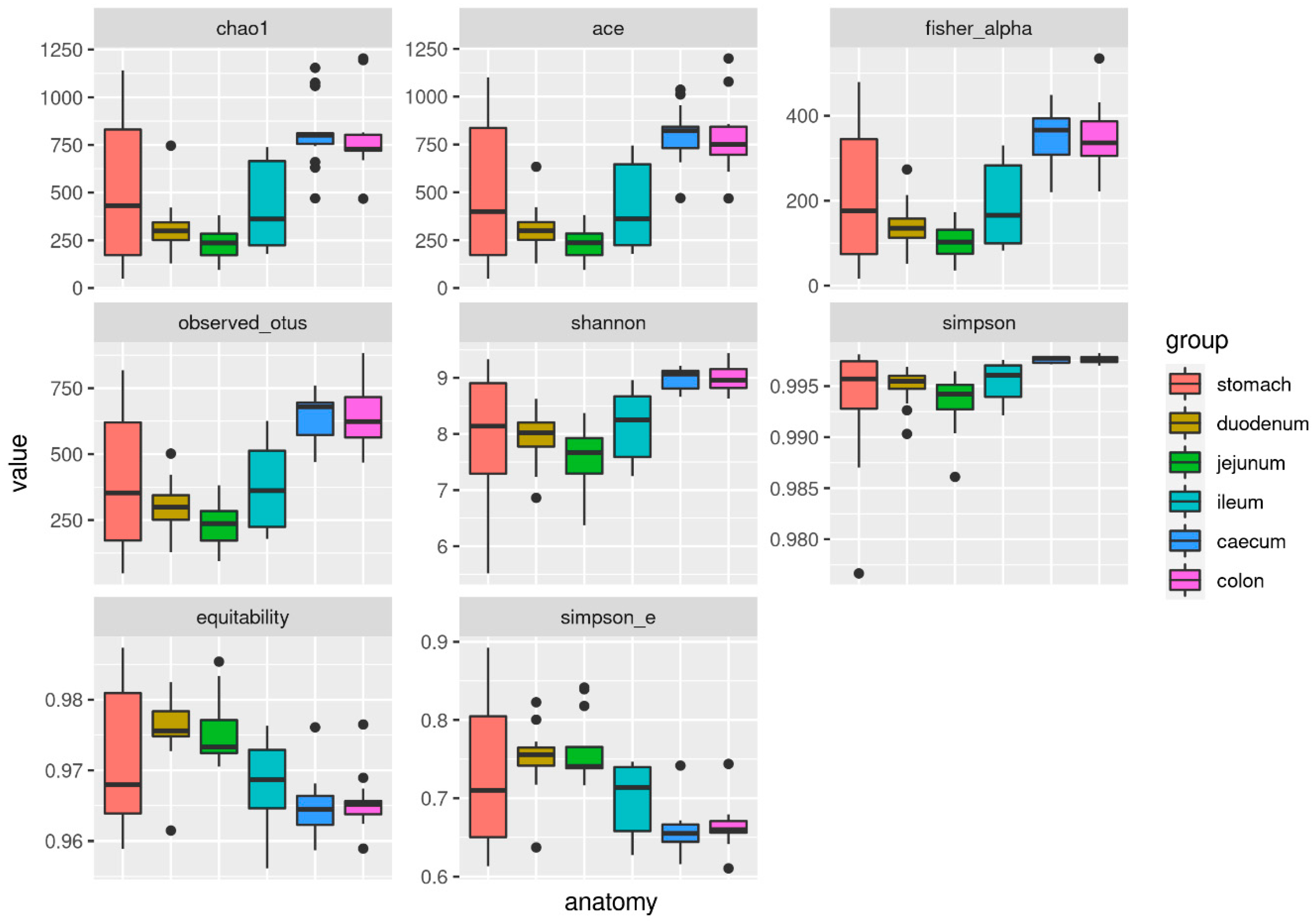
3.4. Alpha and Beta Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liggenstoffer, A.S.; Youssef, N.H.; Couger, M.B.; Elshahed, M.S. Phylogenetic diversity and community structure of anaerobic gut fungi (phylum Neocallimastigomycota) in ruminant and non-ruminant herbivores. ISME J. 2010, 4, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Combes, S.; Michelland, R.J.; Monteils, V.; Cauquil, L.; Soulié, V.; Tran, N.U.; Gidenne, T.; Fortun-Lamothe, L. Postnatal development of the rabbit caecal microbiota composition and activity. FEMS Microbiol. Ecol. 2011, 77, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abecia, L.; Fondevila, M.; Balcells, J.; Edwards, J.E.; Newbold, C.J.; McEwan, N.R. Molecular profiling of bacterial species in the rabbit caecum. FEMS Microbiol. Lett. 2005, 244, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Drouilhet, L.; Achard, C.S.; Zemb, O.; Molette, C.; Gidenne, T.; Larzul, C.; Ruesche, J.; Tircazes, A.; Segura, M.; Bouchez, T.; et al. Direct and correlated responses to selection in two lines of rabbits selected for feed efficiency under ad libitum and restricted feeding: I. Production traits and gut microbiota characteristics. J. Anim. Sci. 2016, 94, 38–48. [Google Scholar] [CrossRef]
- Matthews, C.; Crispie, F.; Lewis, E.; Reid, M.; O’Toole, P.W.; Cotter, P.D. The rumen microbiome: A crucial consideration when optimising milk and meat production and nitrogen utilisation efficiency. Gut Microbes 2019, 10, 115–132. [Google Scholar] [CrossRef]
- Fang, S.; Chen, X.; Zhou, L.; Wang, C.; Chen, Q.; Lin, R.; Xiao, T.; Gan, Q.F. Faecal microbiota and functional capacity associated with weaning weight in meat rabbits. Microb. Biotechnol. 2019, 12, 1441–1452. [Google Scholar] [CrossRef]
- Cremonesi, P.; Ceccarani, C.; Curone, G.; Severgnini, M.; Pollera, C.; Bronzo, V.; Riva, F.; Addis, M.F.; Filipe, J.; Amadori, M.; et al. Milk microbiome diversity and bacterial group prevalence in a comparison between healthy holstein friesian and rendena cows. PLoS ONE 2018, 13, e0205054. [Google Scholar] [CrossRef] [Green Version]
- Zeng, B.; Han, S.; Wang, P.; Wen, B.; Jian, W.; Guo, W.; Yu, Z.; Du, D.; Fu, X.; Kong, F.; et al. The bacterial communities associated with fecal types and body weight of rex rabbits. Sci. Rep. 2015, 5, srep09342. [Google Scholar] [CrossRef]
- Kušar, D.; Avguštin, G. Molecular profiling and identification of methanogenic archaeal species from rabbit caecum. FEMS Microbiol. Ecol. 2010, 74, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Velasco-Galilea, M.; Piles, M.; Viñas, M.; Rafel, O.; González-Rodríguez, O.; Guivernau, M.; Sánchez, J.P. Rabbit microbiota changes throughout the intestinal tract. Front. Microbiol. 2018, 9, 2144. [Google Scholar] [CrossRef] [PubMed]
- Monteils, V.; Cauquil, L.; Combes, S.; Godon, J.J.; Gidenne, T. Potential core species and satellite species in the bacterial community within the rabbit caecum. FEMS Microbiol. Ecol. 2008, 66, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Taraghijou, P.; Safaeiyan, A.; Mobasseri, M.; Ostradrahimi, A. The effect of n-3 long chain fatty acids supplementation on plasma peroxisome proliferator activated. J. Res. Med. Sci. 2013, 17, 942–946. [Google Scholar]
- Li, H.; Qu, J.; Li, T.; Yao, M.; Li, J.; Li, X. Gut microbiota may predict host divergence time during Glires evolution. FEMS Microbiol. Ecol. 2017, 93, fix009. [Google Scholar] [CrossRef] [PubMed]
- Massip, K.; Combes, S.; Cauquil, L.; Zemb, O.; Gidenne, T. High throughput 16S-DNA sequencing for phylogenetic affiliation of the caecal bacterial community in the rabbit—Impact of the hygiene of housing and of intake level. In Proceedings of the INRA-Rowett Symposium on Gut Microbiology, Clermont-Ferrand, France, 17–22 June 2012; p. 57. [Google Scholar]
- Fu, X.; Zeng, B.; Wang, P.; Wang, L.; Wen, B.; Li, Y.; Liu, H.; Bai, S.; Jia, G. Microbiome of total versus live bacteria in the gut of Rex rabbits. Front. Microbiol. 2018, 9, 733. [Google Scholar] [CrossRef]
- Fang, S.; Chen, X.; Ye, X.; Zhou, L.; Xue, S.; Gan, Q. Effects of Gut Microbiome and Short-Chain Fatty Acids (SCFAs) on Finishing Weight of Meat Rabbits. Front. Microbiol. 2020, 11, 1835. [Google Scholar] [CrossRef]
- Zou, F.; Zeng, D.; Wen, B.; Sun, H.; Zhou, Y.; Yang, M.; Peng, Z.; Xu, S.; Wang, H.; Fu, X.; et al. Illumina Miseq platform analysis caecum bacterial communities of rex rabbits fed with different antibiotics. AMB Express 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, M.G.; Rebollar, P.; Mattioli, S.; Castellini, C. n-3 PUFA Sources (Precursor/Products): A Review of Current Knowledge on Rabbit. Animals 2019, 9, 806. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.; Chen, X.; Pan, J.; Chen, Q.; Zhou, L.; Wang, C.; Xiao, T.; Gan, Q.F. Dynamic distribution of gut microbiota in meat rabbits at different growth stages and relationship with average daily gain (ADG). BMC Microbiol. 2020, 20. [Google Scholar] [CrossRef]
- Ericsson, A.C.; Johnson, P.J.; Lopes, M.A.; Perry, S.C.; Lanter, H.R. A microbiological map of the healthy equine gastrointestinal tract. PLoS ONE 2016, 11, e0166523. [Google Scholar] [CrossRef]
- Crowley, E.J.; King, J.M.; Wilkinson, T.; Worgan, H.J.; Huson, K.M.; Rose, M.T.; McEwan, N.R. Comparison of the microbial population in rabbits and guinea pigs by next generation sequencing. PLoS ONE 2017, 12, e0165779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, K.J.; Horng, C.T.; Huang, Y.S.; Hsieh, Y.H.; Wang, C.J.; Yang, J.S.; Lu, C.C.; Chen, F.A. Effects of Lycium barbarum (goji berry) on dry eye disease in rats. Mol. Med. Rep. 2018, 17, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Menchetti, L.; Vecchione, L.; Filipescu, I.; Petrescu, V.F.; Fioretti, B.; Beccari, T.; Ceccarini, M.R.; Codini, M.; Quattrone, A.; Trabalza-Marinucci, M.; et al. Effects of Goji berries supplementation on the productive performance of rabbit. Livest. Sci. 2019, 220, 123–128. [Google Scholar] [CrossRef]
- Castrica, M.; Menchetti, L.; Balzaretti, C.C.M.; Branciari, R.; Ranucci, D.; Cotozzolo, E.; Vigo, D.; Curone, G.; Brecchia, G.; Miraglia, D. Impact of dietary supplementation with goji berries (Lycium barbarum) on microbiological quality, physico-chemical, and sensory characteristics of rabbit meat. Foods 2020, 9, 1480. [Google Scholar] [CrossRef]
- Tait-Burkard, C.; Doeschl-Wilson, A.; McGrew, M.J.; Archibald, A.L.; Sang, H.M.; Houston, R.D.; Whitelaw, C.B.; Watson, M. Livestock 2.0—Genome editing for fitter, healthier, and more productive farmed animals. Genome Biol. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Rinttilä, T.; Apajalahti, J. Intestinal microbiota and metabolites-Implications for broiler chicken health and performance. J. Appl. Poult. Res. 2013, 22, 647–658. [Google Scholar] [CrossRef]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, J.M.D.; Casanova, N.A.; Miyakawa, M.E.F. Microbiota, gut health and chicken productivity: What is the connection? Microorganisms 2019, 7, 374. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Wei, H.; Xu, C.; Xie, X.; Jiang, S.; Peng, J. Maternal soluble fiber diet during pregnancy changes the intestinal microbiota, improves growth performance, and reduces intestinal permeability in piglets. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Zhao, F.; Zhu, W.; Wang, J. Effects of early-life lactoferrin intervention on growth performance, small intestinal function and gut microbiota in suckling piglets. Food Funct. 2019, 10, 5361–5373. [Google Scholar] [CrossRef]
- Boiti, C.; Canali, C.; Brecchia, G.; Zanon, F.; Facchin, E. Effects of induced endometritis on the life-span of corpora lutea in pseudopregnant rabbits and incidence of spontaneous uterine infections related to fertility of breeding does. Theriogenology 1999, 52, 1123–1132. [Google Scholar] [CrossRef]
- Mattioli, S.; Dal Bosco, A.; Combes, S.; Moscati, L.; Crotti, S.; Mancinelli, A.C.; Cotozzolo, E.; Castellini, C. Dehydrated alfalfa and fresh grass supply in young rabbits: Effect on performance and caecal microbiota biodiversity. Animals 2019, 9, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brecchia, G.; Menchetti, L.; Cardinali, R.; Castellini, C.; Polisca, A.; Zerani, M.; Maranesi, M.; Boiti, C. Effects of a bacterial lipopolysaccharide on the reproductive functions of rabbit does. Anim. Reprod. Sci. 2014, 147, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Menchetti, L.; Barbato, O.; Filipescu, I.E.; Traina, G.; Leonardi, L.; Polisca, A.; Troisi, A.; Guelfi, G.; Piro, F.; Brecchia, G. Effects of local lipopolysaccharide administration on the expression of Toll-like receptor 4 and pro-inflammatory cytokines in uterus and oviduct of rabbit does. Theriogenology 2018, 107, 162–174. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Andrew, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 20 November 2020).
- John, J.S. SeqPrep: Tool for Stripping Adaptors and/or Merging Paired Reads with Overlap into Single Reads. 2011. Available online: https://githubcom/jstjohn/SeqPrep (accessed on 20 November 2020).
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucl. Acids 2014, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Weizhong, L.; Adam, G. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar]
- Caporaso, J.G. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Biscarini, F.; Palazzo, F.; Castellani, F.; Masetti, G.; Grotta, L.; Cichelli, A.; Martino, G. Rumen microbiome in dairy calves fed copper and grape-pomace dietary supplementations: Composition and predicted functional profile. PLoS ONE 2018, 13, e0205670. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Chao, A.; Lee, S.M. Estimating the Number of Classes via Sample Coverage. J. Am. Stat. Assoc. 1992, 87, 210. [Google Scholar] [CrossRef]
- Chao, A.; Yang, M.C.K. Stopping rules and estimation for recapture debugging with unequal failure rates. Biometrika 1993, 80, 193–201. [Google Scholar] [CrossRef]
- Shannon, C. Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Fisher, R.A.; Corbet, A.S.; Williams, C.B. The Relation Between the Number of Species and the Number of Individuals in a Random Sample of an Animal Population. J. Anim. Ecol. 1943, 12, 42. [Google Scholar] [CrossRef]
- Smith, B.; Wilson, J.B. A Consumer’s Guide to Evenness Indices. Oikos 1996, 76, 70. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Paulson, J.N.; Colin Stine, O.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016; ISBN 3319242776. [Google Scholar]
- R Core Team R. A Language and Environment for Statistical Computing; R Core Team R: Vienna, Austria, 2018. [Google Scholar]
- Mackie, R.I. Mutualistic fermentative digestion in the gastrointestinal tract: Diversity and evolution. Integr. Comp. Biol. 2002, 42, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Pop, M.; DeBoy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abecia, L.; Rodríguez-Romero, N.; Yañez-Ruiz, D.R.; Fondevila, M. Biodiversity and fermentative activity of caecal microbial communities in wild and farm rabbits from Spain. Anaerobe 2012, 18, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, Y.; Liu, S.; Huang, J.; Zhai, Z.; He, C.; Ding, J.; Wang, J.; Wang, H.; Fan, W.; et al. The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS ONE 2015, 10, e0117441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stearns, J.C.; Lynch, M.D.J.; Senadheera, D.B.; Tenenbaum, H.C.; Goldberg, M.B.; Cvitkovitch, D.G.; Croitoru, K.; Moreno-Hagelsieb, G.; Neufeld, J.D. Bacterial biogeography of the human digestive tract. Sci. Rep. 2011, 1, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Kim, G.B.; Cha, C.J. Spatial heterogeneity and stability of bacterial community in the gastrointestinal tracts of broiler chickens. Poult. Sci. 2014, 93, 1942–1950. [Google Scholar] [CrossRef]
- Li, D.; Chen, H.; Mao, B.; Yang, Q.; Zhao, J.; Gu, Z.; Zhang, H.; Chen, Y.Q.; Chen, W. Microbial Biogeography and Core Microbiota of the Rat Digestive Tract. Sci. Rep. 2017, 8, 45840. [Google Scholar] [CrossRef]
- Bosshard, P.P.; Zbinden, R.; Altwegg, M. Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, Gram-positive bacterium. Int. J. Syst. Evol. Microbiol. 2002, 52, 1263–1266. [Google Scholar] [CrossRef]
- Licht, T.R.; Madsen, B.; Wilcks, A. Selection of bacteria originating from a human intestinal microbiota in the gut of previously germ-free rats. FEMS Microbiol. Lett. 2007, 277, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Falk, A.; Olsson, C.; Ahrné, S.; Molin, G.; Adawi, D.; Jeppsson, B. Ileal pelvic pouch microbiota from two former ulcerative colitis patients, analysed by DNA-based methods, were unstable over time and showed the presence of Clostridium perfringens. Scand. J. Gastroenterol. 2007, 42, 973–985. [Google Scholar] [CrossRef]
- Presley, L.L.; Wei, B.; Braun, J.; Borneman, J. Bacteria associated with immunoregulatory cells in mice. Appl. Environ. Microbiol. 2010, 76, 936–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.H.; Hong, S.H.; Cho, S.B.; Lim, J.S.; Bae, S.E.; Ahn, H.; Lee, E.Y. Characterization of microbial community in the leachate associated with the decomposition of entombed pigs. J. Microbiol. Biotechnol. 2012, 22, 1330–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.C.; Silva, G.; Ramos, R.V.; Staempfli, H.R.; Arroyo, L.G.; Kim, P.; Weese, J.S. Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments in horses. Vet. J. 2015, 205, 74–80. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.M.; Harris, H.M.B.; Jeffery, I.B.; Claesson, M.J.; Younge, B.; O’Toole, P.W.; Ross, R.P. The core faecal bacterial microbiome of Irish Thoroughbred racehorses. Lett. Appl. Microbiol. 2013, 57, 492–501. [Google Scholar] [CrossRef]
- Rajilić-Stojanović, M.; Shanahan, F.; Guarner, F.; De Vos, W.M. Phylogenetic analysis of dysbiosis in ulcerative colitis during remission. Inflamm. Bowel Dis. 2013, 19, 481–488. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, X.; Liu, Y.; Cao, H.; Han, Q.; Xie, B.; Fan, L.; Li, X.; Hu, J.; Yang, G.; et al. Effect of Fermented Corn-Soybean Meal on Serum Immunity, the Expression of Genes Related to Gut Immunity, Gut Microbiota, and Bacterial Metabolites in Grower-Finisher Pigs. Front. Microbiol. 2019, 10, 2620. [Google Scholar] [CrossRef] [Green Version]
- Combes, S.; Gidenne, T.; Cauquil, L.; Bouchez, O.; Fortun-Lamothe, L. Coprophagous behavior of rabbit pups affects implantation of cecal microbiota and health status. J. Anim. Sci. 2014, 92, 652–665. [Google Scholar] [CrossRef]
- Priyadarshini, M.; Kotlo, K.U.; Dudeja, P.K.; Layden, B.T. Role of short chain fatty acid receptors in intestinal physiology and pathophysiology. Compr. Physiol. 2018, 8, 1065–1090. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, C.; Li, F. Impact of dietary fiber/starch ratio in shaping caecal microbiota in rabbits. Can. J. Microbiol. 2015, 61, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The firmicutes/bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Stomach | Duodenum | Jejunum | Ileum | Caecum | Colon | p-Value |
|---|---|---|---|---|---|---|---|
| Firmicutes | 0.68 ± 0.27 | 0.40 ± 0.23 | 0.41 ± 0.26 | 0.40 ± 0.19 | 0.43 ± 0.08 | 0.44 ± 0.07 | 0.00 |
| Bacteroidetes | 0.16 ± 0.16 | 0.02 ± 0.03 | 0.01 ± 0.02 | 0.16 ± 0.16 | 0.40 ± 0.06 | 0.38 ± 0.08 | 0.00 |
| Verrucomicrobia | 0.05 ± 0.05 | 0.04 ± 0.04 | 0.02 ± 0.02 | 0.08 ± 0.07 | 0.15 ± 0.04 | 0.14 ± 0.03 | 0.00 |
| Patescibacteria | 0.03 ± 0.02 | 0.13 ± 0.11 | 0.14 ± 0.13 | 0.10 ± 0.11 | 0.01 ± 0.00 | 0.01 ± 0.01 | 0.00 |
| Euryarchaeota | 0.06 ± 0.09 | 0.31 ± 0.26 | 0.30 ± 0.28 | 0.18 ± 0.23 | 0.01 ± 0.01 | 0.02 ± 0.02 | 0.00 |
| Actinobacteria | 0.02 ± 0.03 | 0.09 ± 0.16 | 0.13 ± 0.22 | 0.09 ± 0.18 | 0.00 ± 0.01 | 0.00 ± 0.00 | 0.06 |
| Cyanobacteria | 0.01 ± 0.01 | 0.01 ± 0.01 | 0.00 ± 0.01 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.14 |
| Proteobacteria | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.02 |
| Tenericutes | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.59 |
| Epsilonbacteraeota | 0.00 ± 0.00 | 0.00 ± 0.01 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.13 |
| Lentisphaerae | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.85 |
| Synergistetes | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 |
| Metric | Stomach | Duodenum | Jejunum | Ileum | Caecum | Colon | p-Value |
|---|---|---|---|---|---|---|---|
| Chao1 | 508.51 ± 389.20 | 316.86 ± 148.15 | 233.50 ± 85.33 | 432.19 ± 220.60 | 814.53 ± 181.13 | 787.36 ± 192.90 | 0.00 |
| Ace | 493.36 ± 376.40 | 308.82 ± 124.18 | 233.50 ± 85.33 | 424.02 ± 214.95 | 803.85 ± 147.50 | 785.11 ± 181.83 | 0.00 |
| Fisher_alpha | 210.88 ± 163.59 | 139.61 ± 57.78 | 102.52 ± 40.79 | 187.01 ± 94.71 | 354.15 ± 60.28 | 348.24 ± 78.62 | 0.00 |
| Observed otus | 399.71 ± 271.22 | 299.43 ± 100.43 | 233.50 ± 85.33 | 374.86 ± 160.72 | 645.43 ± 84.73 | 638.79 ± 111.72 | 0.00 |
| Shannon | 7.97 ± 1.17 | 7.94 ± 0.50 | 7.58 ± 0.56 | 8.15 ± 0.60 | 8.99 ± 0.18 | 8.98 ± 0.23 | 0.00 |
| Simpson | 0.99 ± 0.01 | 1.00 ± 0.00 | 0.99 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | 0.00 |
| Equitability | 0.97 ± 0.01 | 0.98 ± 0.00 | 0.98 ± 0.00 | 0.97 ± 0.01 | 0.96 ± 0.00 | 0.97 ± 0.00 | 0.00 |
| Simpson e | 0.73 ± 0.10 | 0.75 ± 0.04 | 0.76 ± 0.04 | 0.70 ± 0.04 | 0.66 ± 0.03 | 0.66 ± 0.03 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cotozzolo, E.; Cremonesi, P.; Curone, G.; Menchetti, L.; Riva, F.; Biscarini, F.; Marongiu, M.L.; Castrica, M.; Castiglioni, B.; Miraglia, D.; et al. Characterization of Bacterial Microbiota Composition along the Gastrointestinal Tract in Rabbits. Animals 2021, 11, 31. https://doi.org/10.3390/ani11010031
Cotozzolo E, Cremonesi P, Curone G, Menchetti L, Riva F, Biscarini F, Marongiu ML, Castrica M, Castiglioni B, Miraglia D, et al. Characterization of Bacterial Microbiota Composition along the Gastrointestinal Tract in Rabbits. Animals. 2021; 11(1):31. https://doi.org/10.3390/ani11010031
Chicago/Turabian StyleCotozzolo, Elisa, Paola Cremonesi, Giulio Curone, Laura Menchetti, Federica Riva, Filippo Biscarini, Maria Laura Marongiu, Marta Castrica, Bianca Castiglioni, Dino Miraglia, and et al. 2021. "Characterization of Bacterial Microbiota Composition along the Gastrointestinal Tract in Rabbits" Animals 11, no. 1: 31. https://doi.org/10.3390/ani11010031
APA StyleCotozzolo, E., Cremonesi, P., Curone, G., Menchetti, L., Riva, F., Biscarini, F., Marongiu, M. L., Castrica, M., Castiglioni, B., Miraglia, D., Luridiana, S., & Brecchia, G. (2021). Characterization of Bacterial Microbiota Composition along the Gastrointestinal Tract in Rabbits. Animals, 11(1), 31. https://doi.org/10.3390/ani11010031