Effects of Multi-Species Direct-Fed Microbial Products on Ruminal Metatranscriptome and Carboxyl-Metabolome of Beef Steers

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Rumen Fluid Sampling
2.2. Metatranscriptomics Analysis
2.3. CIL-LC/MS-Based Metabolomics Analysis
2.4. Data and Statistical Analysis
2.4.1. Metatranscriptomics Analysis
2.4.2. Metabolomics Data Analysis
3. Results
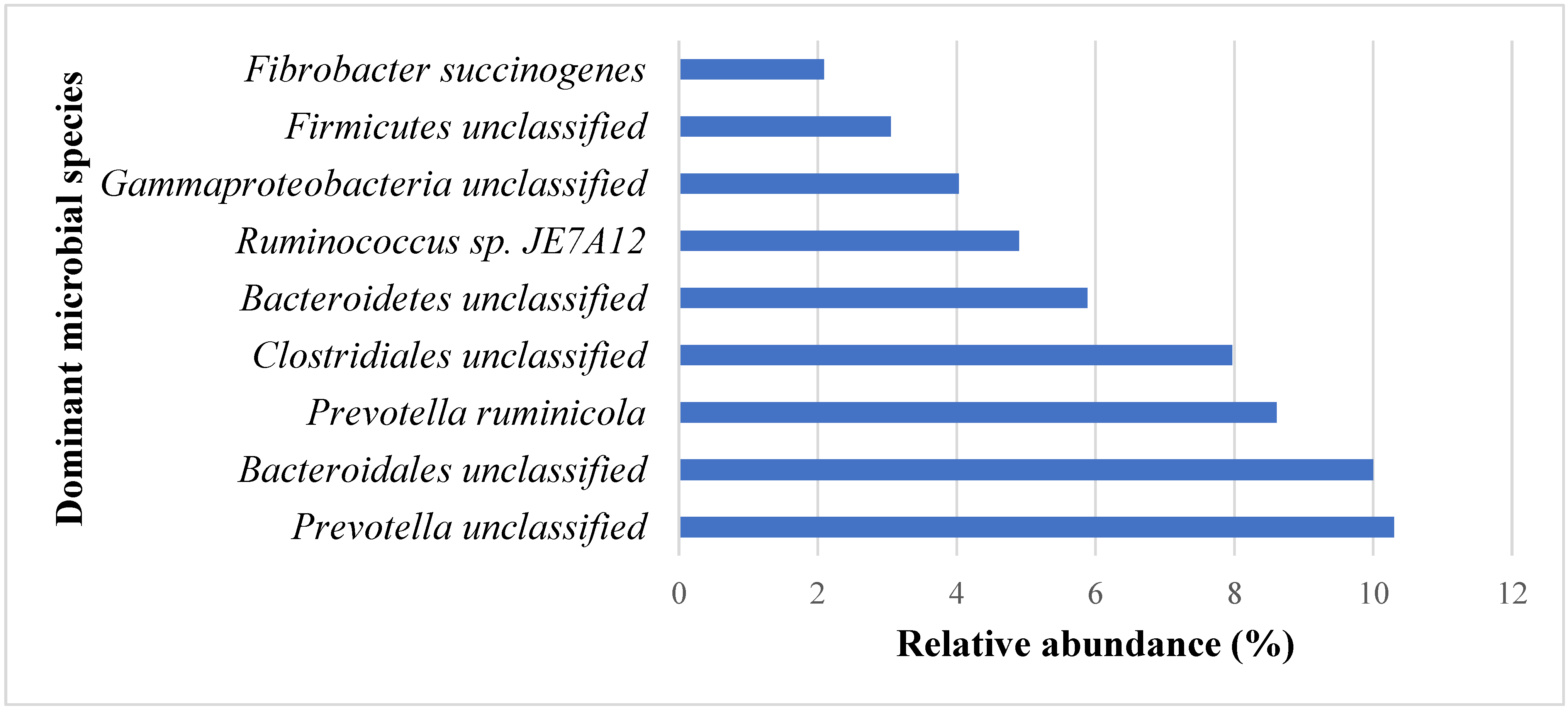
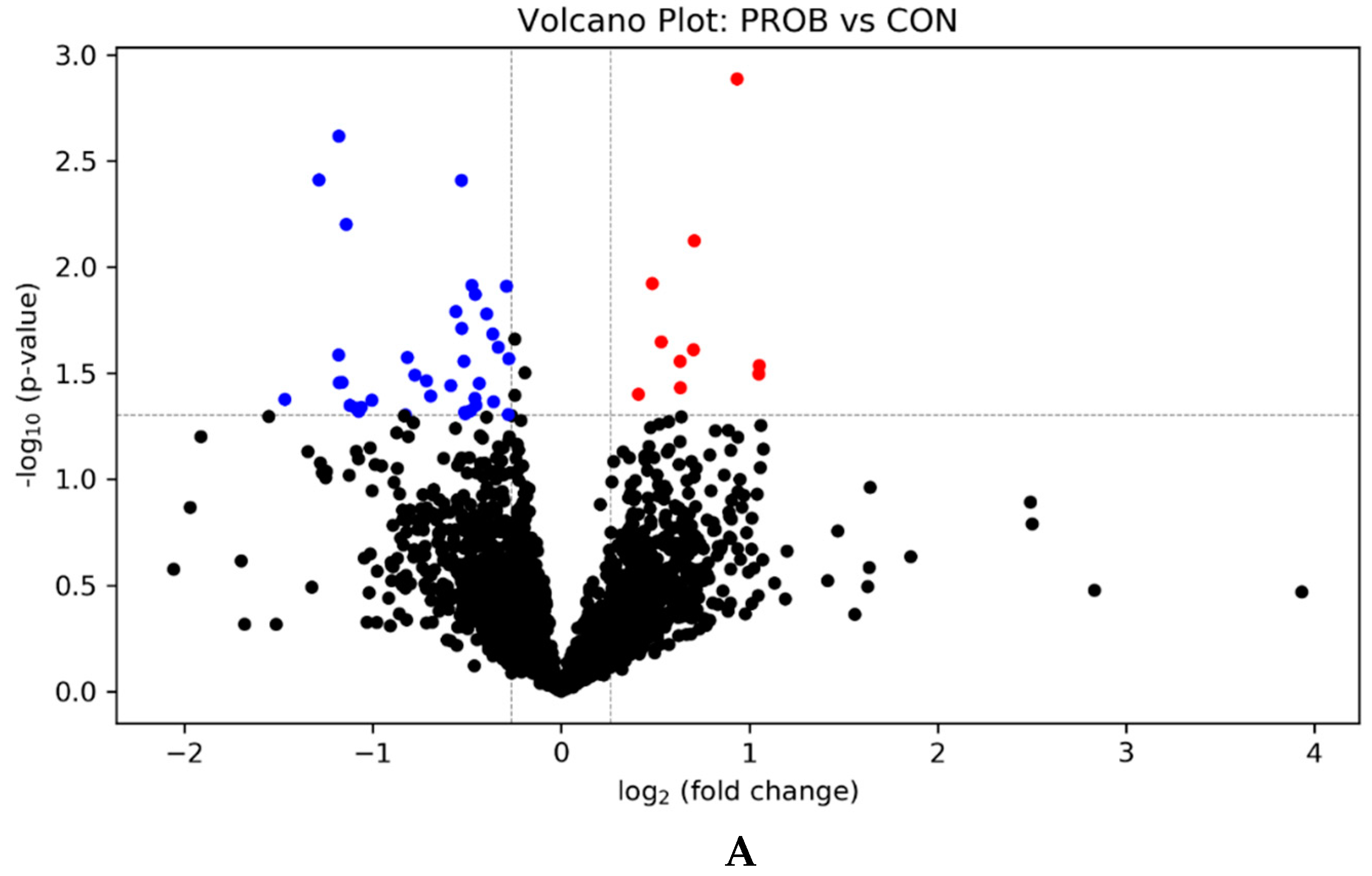
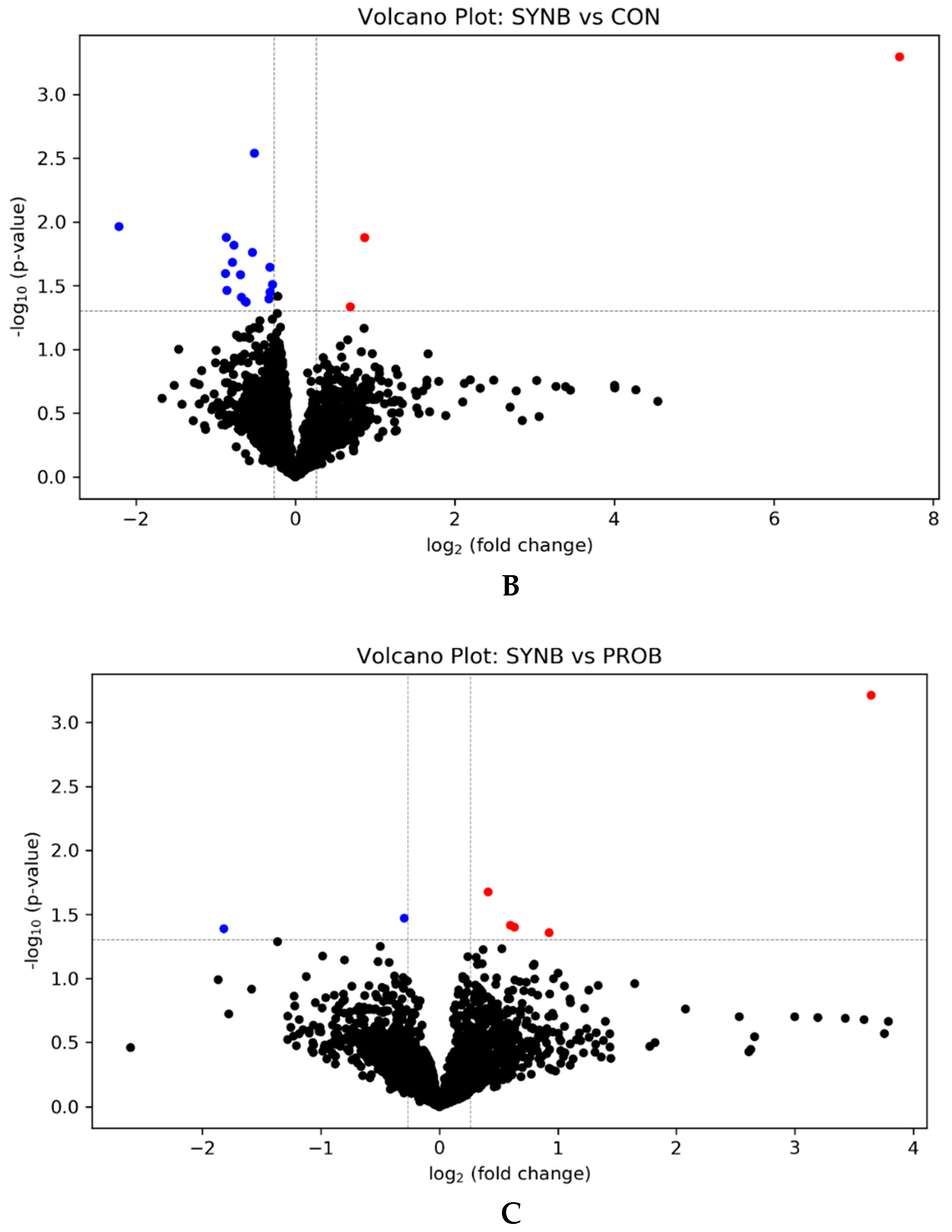
3.1. Effects of PROB and SYNB on Ruminal Metatranscriptome Profile
3.2. Effects of PROB and SYNB on Ruminal Carboxyl-Metabolome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krehbiel, C.R.; Rust, S.R.; Zhang, G.; Gilliland, S.E. Bacterial direct-fed microbials in ruminant diets: Performance response and mode of action. J. Anim. Sci. 2003, 81, E120–E132. [Google Scholar]
- McAllister, T.A.; Beauchemin, K.A.; Alazzeh, A.Y.; Baah, J.; Teather, R.M.; Stanford, K. Review: The use of direct fed microbials to mitigate pathogens and enhance production in cattle. Can. J. Anim. Sci. 2011, 91, 193–211. [Google Scholar] [CrossRef] [Green Version]
- Fomenky, B.E.; Do, D.N.; Talbot, G.; Chiquette, J.; Bissonnette, N.; Chouinard, Y.P.; Lessard, M.; Ibeagha-Awemu, E.M. Direct-fed microbial supplementation influences the bacteria community composition of the gastrointestinal tract of pre- and post-weaned calves. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Ogunade, I.M.; Lay, J.; Andries, K.; McManus, C.J.; Bebe, F. Effects of live yeast on differential genetic and functional attributes of rumen microbiota in beef cattle. J. Anim. Sci. Biotechnol. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Ogunade, I.; Qi, S.; Hackmann, T.; Staples, C.; Adesogan, A. Effects of the dose and viability of Saccharomyces cerevisiae. 1. Diversity of ruminal microbes as analyzed by Illumina MiSeq sequencing and quantitative PCR. J. Dairy Sci. 2017, 100, 325–342. [Google Scholar] [CrossRef] [Green Version]
- Guo, K.; Li, L. High-Performance Isotope Labeling for Profiling Carboxylic Acid-Containing Metabolites in Biofluids by Mass Spectrometry. Anal. Chem. 2010, 82, 8789–8793. [Google Scholar] [CrossRef]
- Ogunade, I.M.; McCoun, M.; Idowu, M.D.; Peters, S.O. Comparative effects of two multispecies direct-fed microbial products on energy status, nutrient digestibility, and ruminal fermentation, bacterial community, and metabolome of beef steers. J. Anim. Sci. 2020, 98, skaa201. [Google Scholar] [CrossRef]
- Hristov, A.N.; Callaway, T.R.; Lee, C.; Dowd, S.E. Rumen bacterial, archaeal, and fungal diversity of dairy cows in response to ingestion of lauric or myristic acid1. J. Anim. Sci. 2012, 90, 4449–4457. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Dawe, M.; Guo, K.; Li, L. Development of High-Performance Chemical Isotope Labeling LC–MS for Profiling the Carbonyl Submetabolome. Anal. Chem. 2017, 89, 6758–6765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Li, L. Determination of Total Concentration of Chemically Labeled Metabolites as a Means of Metabolome Sample Normalization and Sample Loading Optimization in Mass Spectrometry-Based Metabolomics. Anal. Chem. 2012, 84, 10723–10731. [Google Scholar] [CrossRef] [PubMed]
- Mung, D.; Li, L. Development of Chemical Isotope Labeling LC-MS for Milk Metabolomics: Comprehensive and Quantitative Profiling of the Amine/Phenol Submetabolome. Anal. Chem. 2017, 89, 4435–4443. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Huan, T.; Li, L. Quantitative Metabolome Analysis Based on Chromatographic Peak Reconstruction in Chemical Isotope Labeling Liquid Chromatography Mass Spectrometry. Anal. Chem. 2015, 87, 7011–7016. [Google Scholar] [CrossRef]
- Li, L.; Li, R.; Zhou, J.; Zuniga, A.; Stanislaus, A.E.; Wu, Y.; Huan, T.; Zheng, J.; Shi, Y.; Wishart, D.S.; et al. MyCompoundID: Using an Evidence-Based Metabolome Library for Metabolite Identification. Anal. Chem. 2013, 85, 3401–3408. [Google Scholar] [CrossRef]
- Gibson, G.R.; Probert, H.M.; Van Loo, J.; Rastall, R.A.; Roberfroid, M.B. Dietary modulation of the human colonic microbiota: Updating the concept of prebiotics. Nutr. Res. Rev. 2004, 17, 259–275. [Google Scholar] [CrossRef] [Green Version]
- Taxis, T.M.; Wolff, S.; Gregg, S.J.; Minton, N.O.; Zhang, C.; Dai, J.; Schnabel, R.D.; Taylor, J.F.; Kerley, M.S.; Pires, J.C.; et al. The players may change but the game remains: Network analyses of ruminal microbiomes suggest taxonomic differences mask functional similarity. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekele, A.Z.; Koike, S.; Kobayashi, Y. Genetic diversity and diet specificity of ruminal Prevotella revealed by 16S rRNA gene-based analysis. FEMS Microbiol. Lett. 2010, 305, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Pitta, D.W.; Pinchak, W.E.; Dowd, S.E.; Osterstock, J.; Gontcharova, V.; Youn, E.; Dorton, K.; Yoon, I.; Min, B.R.; Fulford, J.D.; et al. Rumen Bacterial Diversity Dynamics Associated with Changing from Bermudagrass Hay to Grazed Winter Wheat Diets. Microb. Ecol. 2009, 59, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.M.; Weimer, P.J. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl. Microbiol. Biotechnol. 2007, 75, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Lettat, A.; Hassanat, F.; Benchaar, C. Corn silage in dairy cow diets to reduce ruminal methanogenesis: Effects on the rumen metabolically active microbial communities. J. Dairy Sci. 2013, 96, 5237–5248. [Google Scholar] [CrossRef]
- Smedley, J.G.; Fisher, D.J.; Sayeed, S.; Chakrabarti, G.; McClane, B.A. The Enteric Toxins of Clostridium Perfringens; Springer: Berlin/Heidelberg, Germany, 2004; pp. 183–204. [Google Scholar]
- Stephens, T.P.; Loneragan, G.H.; Karunasena, E.; Brashears, M.M. Reduction of Escherichia coli O157 and Salmonella in Feces and on Hides of Feedlot Cattle Using Various Doses of a Direct-Fed Microbial. J. Food Prot. 2007, 70, 2386–2391. [Google Scholar] [CrossRef]
- Tabe, E.S.; Oloya, J.; Doetkott, D.K.; Bauer, M.L.; Gibbs, P.S.; Khaitsa, M.L. Comparative Effect of Direct-Fed Microbials on Fecal Shedding of Escherichia coli O157:H7 and Salmonella in Naturally Infected Feedlot Cattle. J. Food Prot. 2008, 71, 539–544. [Google Scholar] [CrossRef]
- Callaway, T.R.; Anderson, R.C.; Edrington, T.S.; Genovese, K.J.; Bischoff, K.M.; Poole, T.L.; Jung, Y.S.; Harvey, R.B.; Nisbet, D.J. What are we doing about Escherichia coli O157:H7 in cattle? J. Anim. Sci. 2004, 82, E93–E99. [Google Scholar] [CrossRef]
- Chaucheyras-Durand, F.; Fonty, G. Influence of a Probiotic Yeast ( Saccharomyces cerevisiae CNCM I-1077) on Microbial Colonization and Fermentations in the Rumen of Newborn Lambs. Microb. Ecol. Health Dis. 2002, 14, 30–36. [Google Scholar] [CrossRef]
- Newbold, C.J.; Wallace, R.J.; Mcintosh, F.M. Mode of action of the yeast Saccharomyces cerevisiae as a feed additive for ruminants. Br. J. Nutr. 1996, 76, 249. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Hitch, T.C.; Chen, Y.; Creevey, C.J. Comparative metagenomic and metatranscriptomic analyses reveal the breed effect on the rumen microbiome and its associations with feed efficiency in beef cattle. Microbiome 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roehe, R.; Dewhurst, R.J.; Duthie, C.A.; Rooke, J.A.; McKain, N.; Ross, D.W.; Hyslop, J.J.; Waterhouse, A.; Freeman, T.C.; Watson, M.; et al. Bovine Host Genetic Variation Influences Rumen Microbial Methane Production with Best Selection Criterion for Low Methane Emitting and Efficiently Feed Converting Hosts Based on Metagenomic Gene Abundance. PLoS Genet. 2016, 12, e1005846. [Google Scholar] [CrossRef] [PubMed]
- Kiran, R.R.; Kumar, D.S. Influence of yeast culture supplementation on rumen fermentation of bulls fed complete rations. Int. J. Agric. Sci. Vet. Med. 2013, 1, 8–15. [Google Scholar]
- Mao, S.Y.; Zhang, R.Y.; Wang, D.S.; Zhut, W.Y. Impact of subacute ruminal acidosis (SARA) adaptation on rumen microbiota in dairy cattle using pyrosequencing. Anaerobe 2013, 24, 12–19. [Google Scholar] [CrossRef]
- Ueki, A.; Akasaka, H.; Satoh, A.; Suzuki, D.; Ueki, K. Prevotella paludivivens sp. nov., a novel strictly anaerobic, Gram-negative, hemicellulose-decomposing bacterium isolated from plant residue and rice roots in irrigated rice-field soil. Int. J. Syst. Evol. Microbiol. 2007, 57, 1803–1809. [Google Scholar] [CrossRef]
- White, W.H.; Gunyuzlu, P.L.; Toyn, J.H. Saccharomyces cerevisiaeIs Capable ofde NovoPantothenic Acid Biosynthesis Involving a Novel Pathway of β-Alanine Production from Spermine. J. Biol. Chem. 2001, 276, 10794–10800. [Google Scholar] [CrossRef] [Green Version]
- Wirth, R.; Kádár, G.; Kakuk, B.; Maróti, G.; Bagi, Z.; Szilágyi, Á.; Rákhely, G.; Horváth, J.; Kovács, K.L. The planktonic core microbiome and core functions in the cattle rumen by next generation sequencing. Front. Microbiol. 2018, 9, 2285. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Jin, Y.; He, M.L.; Li, C.; Beauchemin, K.A.; Yang, W.Z. Effects of increasing levels of corn dried distillers grains with solubles and monensin on ruminal biohydrogenation and duodenal flows of fatty acids in beef heifers fed high-grain diets1. J. Anim. Sci. 2014, 92, 1089–1098. [Google Scholar] [CrossRef]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef]
- Ellis, J.E.; Williams, A.G.; Lloyd, D. Oxygen consumption by ruminal microorganisms: Protozoal and bacterial contributions. Appl. Environ. Microbiol. 1989, 55, 2583–2587. [Google Scholar] [CrossRef] [Green Version]
- Ren, R.; Hashimoto, T.; Mizuno, M.; Takigawa, H.; Yoshida, M.; Azuma, T.; Kanazawa, K. A lipid peroxidation product 9-oxononanoic acid induces phospholipase A2 activity and thromboxane A2 production in human blood. J. Clin. Biochem. Nutr. 2013, 52, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Wei, P.; Duke, R.W.; Reaven, P.D.; Harman, S.M.; Cutler, R.G.; Heward, C.B. Quantification of 8-iso-prostaglandin-F2α and 2,3-dinor-8-iso-prostaglandin-F2α in human urine using liquid chromatography-tandem mass spectrometry. Free Radic. Biol. Med. 2003, 34, 409–418. [Google Scholar] [CrossRef]
- Chaucheyras-Durand, F.; Walker, N.D.; Bach, A. Effects of active dry yeasts on the rumen microbial ecosystem: Past, present and future. Anim. Feed Sci. Technol. 2008, 145, 5–26. [Google Scholar] [CrossRef]
- Ogunade, I.; Schweickart, H.; McCoun, M.; Cannon, K.; McManus, C. Integrating 16S rRNA sequencing and LC–MS-based metabolomics to evaluate the effects of live yeast on rumen function in beef cattle. Animals 2019, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beattie, D.S.; Jenkins, H.C.; Howton, M.M. Biochemical Evidence for the Orientation of Cytochrome b in the Yeast Mitochondrial Membrane in the Eight-Helix Model. Arch. Biochem. Biophys. 1994, 312, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Chaturvedi, J.; Chaudhari, B.P.; Singh, R.L.; Kakkar, P. Probiotic Enterococcus lactis IITRHR1 protects against acetaminophen-induced hepatotoxicity. Nutrition 2012, 28, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, R.; Wang, L.; Zhang, H. The antioxidative effects of probiotic Lactobacillus casei Zhang on the hyperlipidemic rats. Eur. Food Res. Technol. 2010, 231, 151–158. [Google Scholar] [CrossRef]
- Sun, J.; Hu, X.L.; Le, G.W.; Shi, Y.H. Lactobacilli prevent hydroxy radical production and inhibit Escherichia coli and Enterococcus growth in system mimicking colon fermentation. Lett. Appl. Microbiol. 2010, 50, 264–269. [Google Scholar] [CrossRef]
- Uchida, K. Histidine and lysine as targets of oxidative modification. Amino Acids 2003, 25, 249–257. [Google Scholar] [CrossRef]
- Requena, J.R.; Chao, C.C.; Levine, R.L.; Stadtman, E.R. Glutamic and aminoadipic semialdehydes are the main carbonyl products of metal-catalyzed oxidation of proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 69–74. [Google Scholar] [CrossRef]
- Gao, X.; Liang, M.; Fang, Y.; Zhao, F.; Tian, J.; Zhang, X.; Qin, X. Deciphering the differential effective and toxic responses of bupleuri radix following the induction of chronic unpredictable mild stress and in healthy rats based on serum metabolic profiles. Front. Pharmacol. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient (%DM) | % of Dietary DM |
|---|---|
| Corn silage | 79.7 |
| Dehydrated distillers grain | 9.06 |
| Soybean meal | 9.28 |
| Limestone | 0.42 |
| Deccox 2 | 0.03 |
| Vitamin and mineral premix 3 | 1.51 |
| Nutrient analysis 4 | |
| DM, % | 44.5 |
| CP, % | 14.7 |
| aNDF, % | 38.6 |
| ADF, % | 21.5 |
| EE, % | 3.50 |
| Ca, % | 0.87 |
| P, % | 0.63 |
| TDN, % | 72.6 |
| NEm, Mcal/kg | 1.72 |
| NEg, Mcal/kg | 1.10 |
| Item | Normalized RT | FC | FDR |
|---|---|---|---|
| Effects of supplemental PROB | |||
| 2-amino-5-oxohexanoate | 442.4 | 0.41 | 0.01 |
| 6-acetamido-2-oxohexanoate | 638.7 | 0.45 | 0.01 |
| 9-oxononanoic acid | 1256.5 | 0.69 | 0.02 |
| Isomer of 9-oxononanoic acid * | 1097.7 | 0.74 | 0.04 |
| 8-iso prostaglandin F1alpha | 1245.5 | 0.82 | 0.01 |
| 9,10-epoxy-13-hydroxy-11-octadecenoate | 1330.5 | 0.70 | 0.05 |
| 12,13-epoxy-9-hydroxy-10-octadecenoate | 1337.4 | 0.70 | 0.05 |
| Isomer of 12,13-epoxy-9-hydroxy-10-octadecenoate * | 1364.4 | 0.72 | 0.05 |
| Succinic acid | 973.7 | 0.61 | 0.03 |
| Hydroxylpropionic acid | 424.9 | 1.52 | 0.01 |
| Propionic acid | 758.4 | 1.43 | 0.05 |
| Isomer of propionic acid | 723.7 | 1.71 | 0.01 |
| Effects of supplemental SYNB | |||
| Succinic acid | 973.7 | 0.74 | 0.01 |
| Pimelate | 1374.2 | 0.54 | 0.03 |
| Isomer of propionic acid | 723.7 | 1.32 | 0.05 |
| Propionic acid | 758.4 | 1.54 | 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCoun, M.; Oyebade, A.; Estrada-Reyes, Z.M.; Pech-Cervantes, A.A.; Ogunade, I.M. Effects of Multi-Species Direct-Fed Microbial Products on Ruminal Metatranscriptome and Carboxyl-Metabolome of Beef Steers. Animals 2021, 11, 72. https://doi.org/10.3390/ani11010072
McCoun M, Oyebade A, Estrada-Reyes ZM, Pech-Cervantes AA, Ogunade IM. Effects of Multi-Species Direct-Fed Microbial Products on Ruminal Metatranscriptome and Carboxyl-Metabolome of Beef Steers. Animals. 2021; 11(1):72. https://doi.org/10.3390/ani11010072
Chicago/Turabian StyleMcCoun, Megan, Adeoye Oyebade, Zaira M. Estrada-Reyes, Andres A. Pech-Cervantes, and Ibukun M. Ogunade. 2021. "Effects of Multi-Species Direct-Fed Microbial Products on Ruminal Metatranscriptome and Carboxyl-Metabolome of Beef Steers" Animals 11, no. 1: 72. https://doi.org/10.3390/ani11010072
APA StyleMcCoun, M., Oyebade, A., Estrada-Reyes, Z. M., Pech-Cervantes, A. A., & Ogunade, I. M. (2021). Effects of Multi-Species Direct-Fed Microbial Products on Ruminal Metatranscriptome and Carboxyl-Metabolome of Beef Steers. Animals, 11(1), 72. https://doi.org/10.3390/ani11010072