
Assessment and Distribution of Runs of Homozygosity in Horse Breeds Representing Different Utility Types

 , , , , , ,
, , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Material, DNA Isolation and Genotyping, Filtration of Genotypic Data
2.2. Runs of Homozygosity Identification and Estimation of FROH
2.3. Identification of ROH Islands and Regions of No ROH Presence
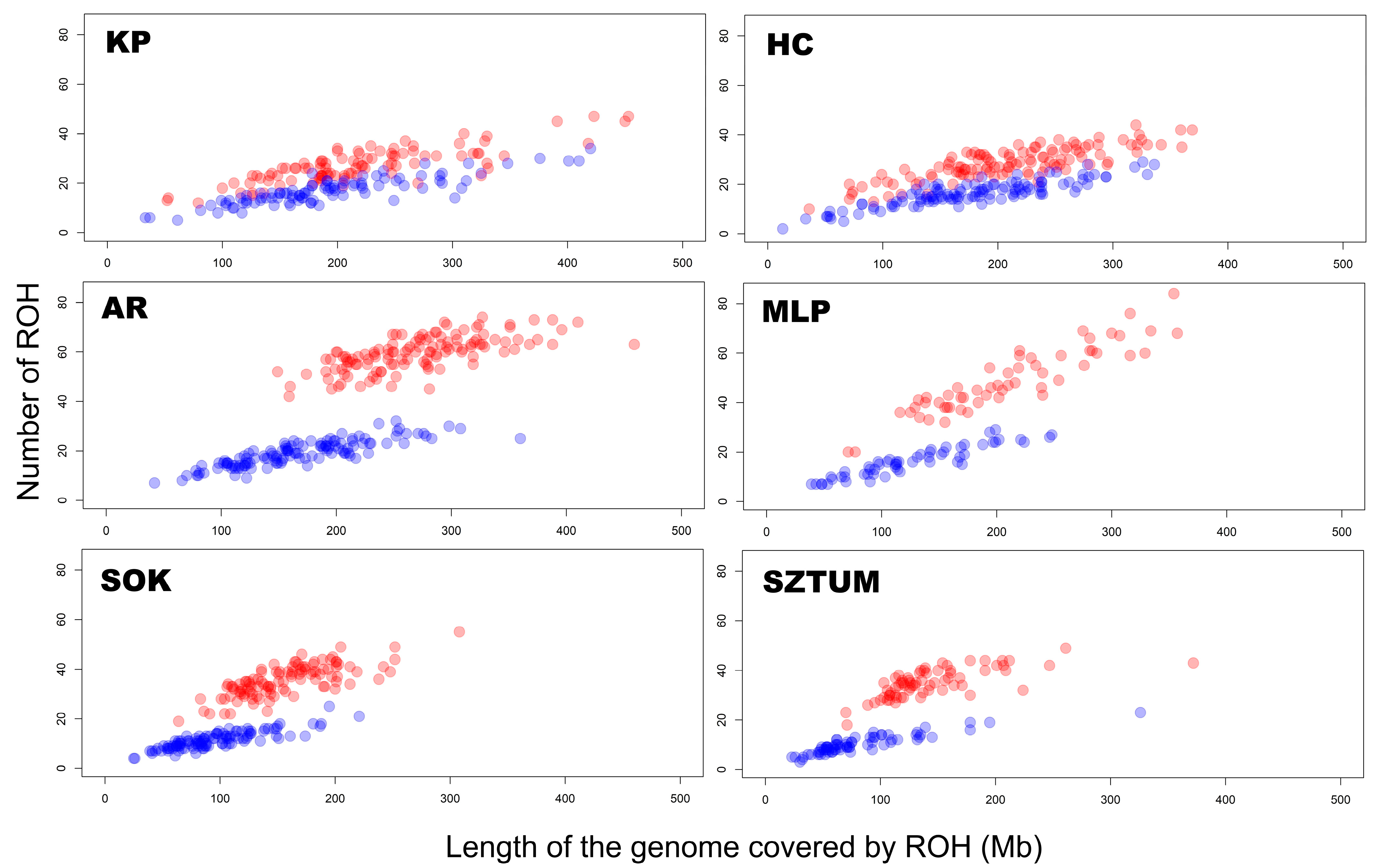
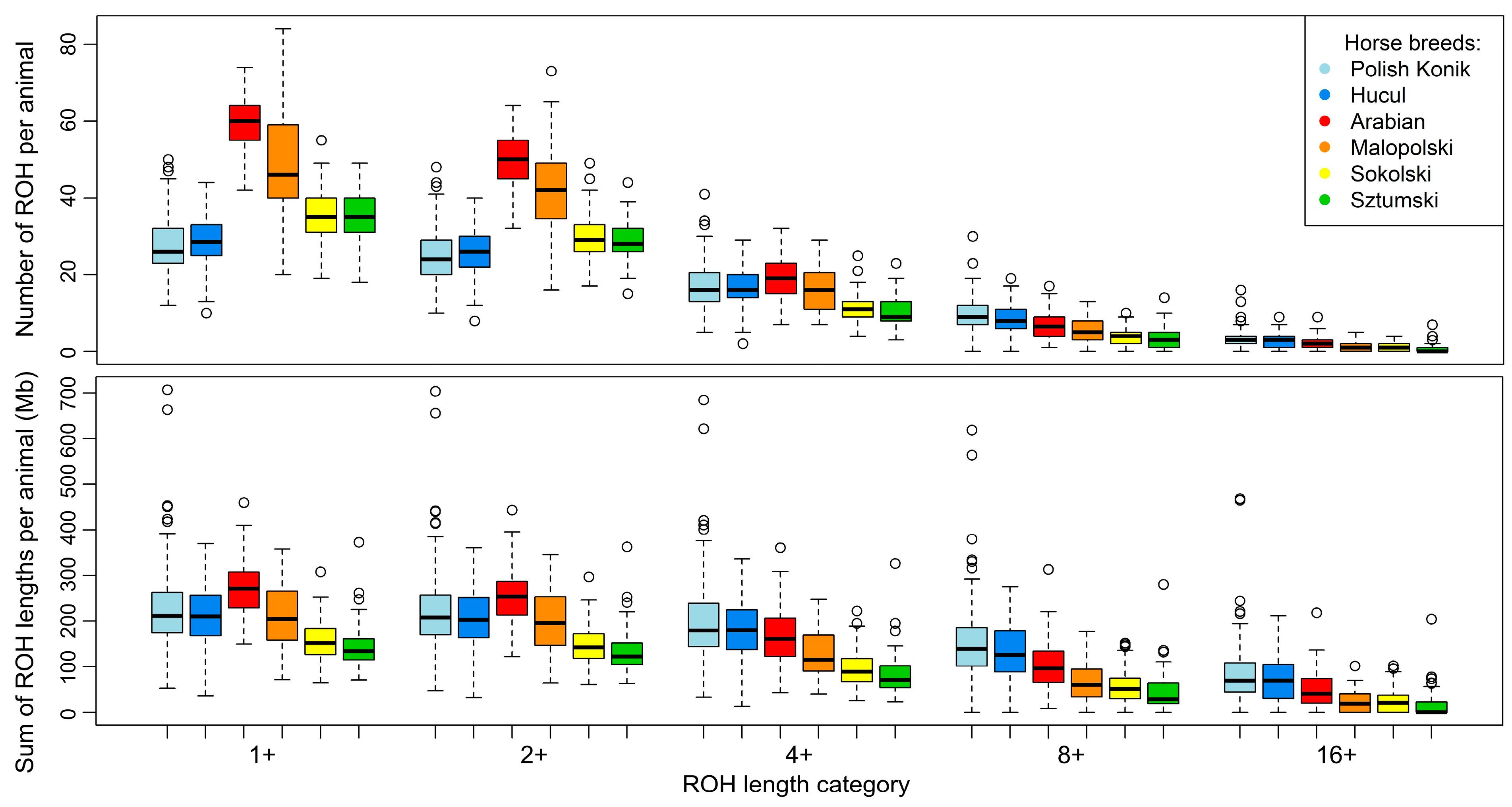
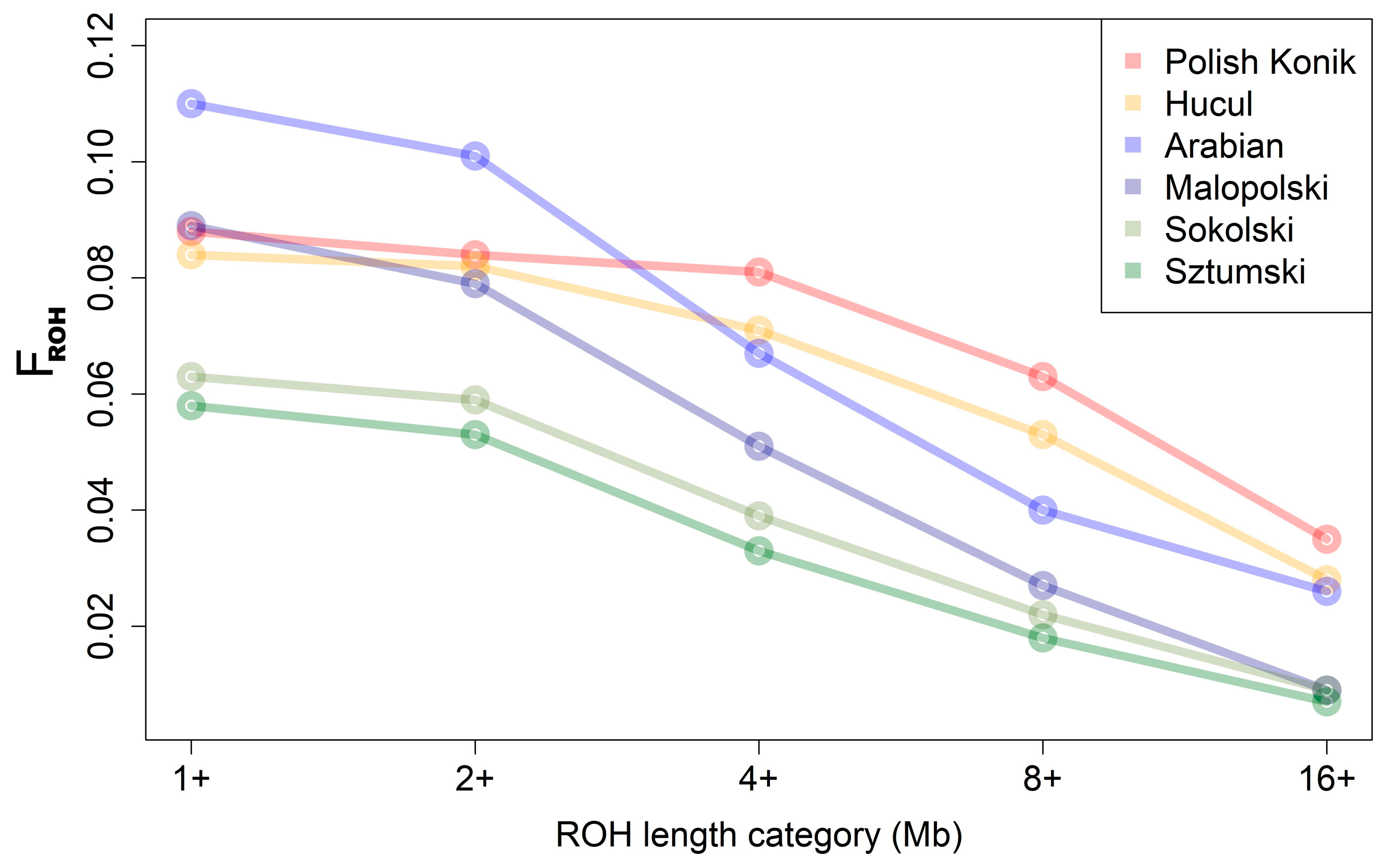
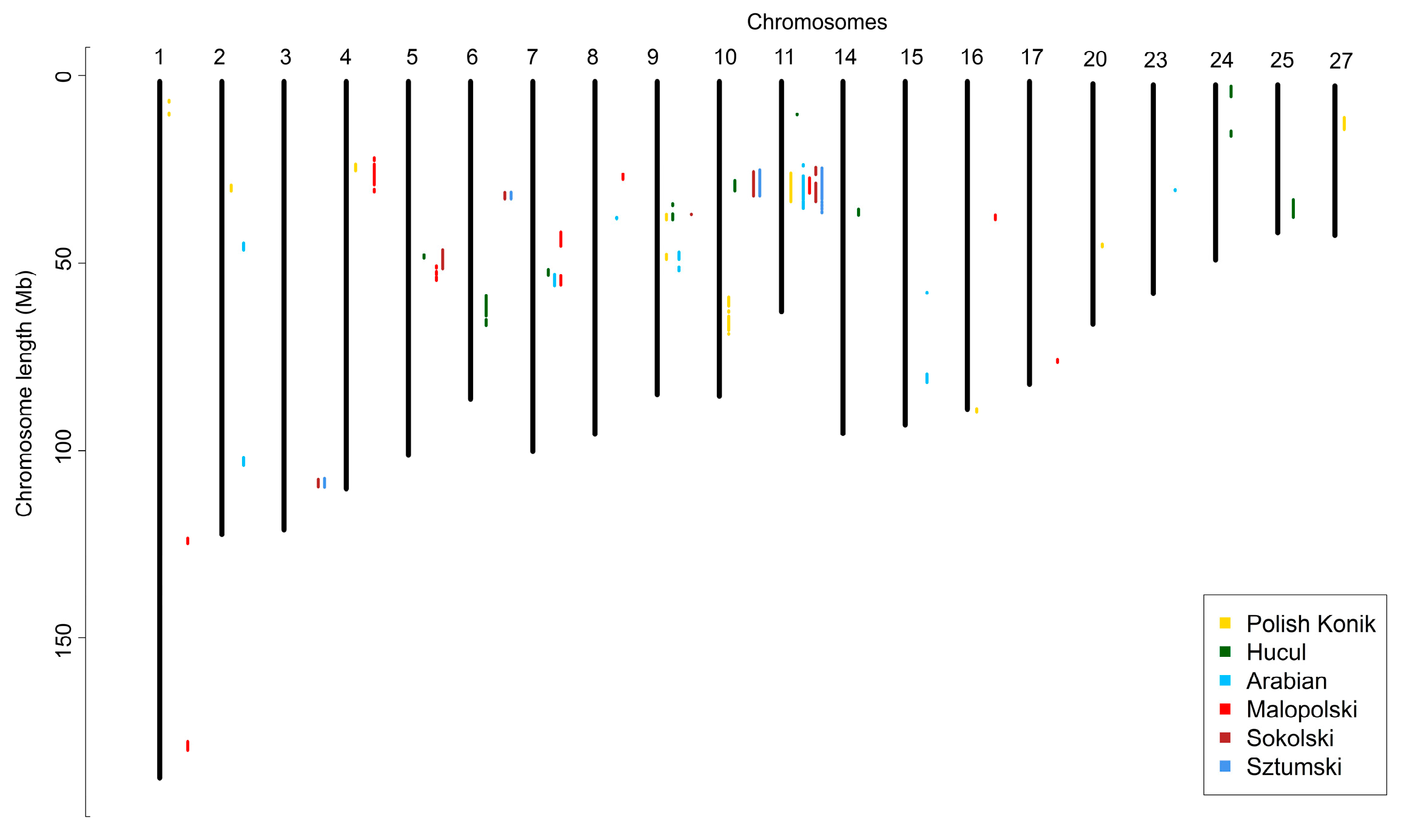
3. Results
3.1. ROH Characteristics
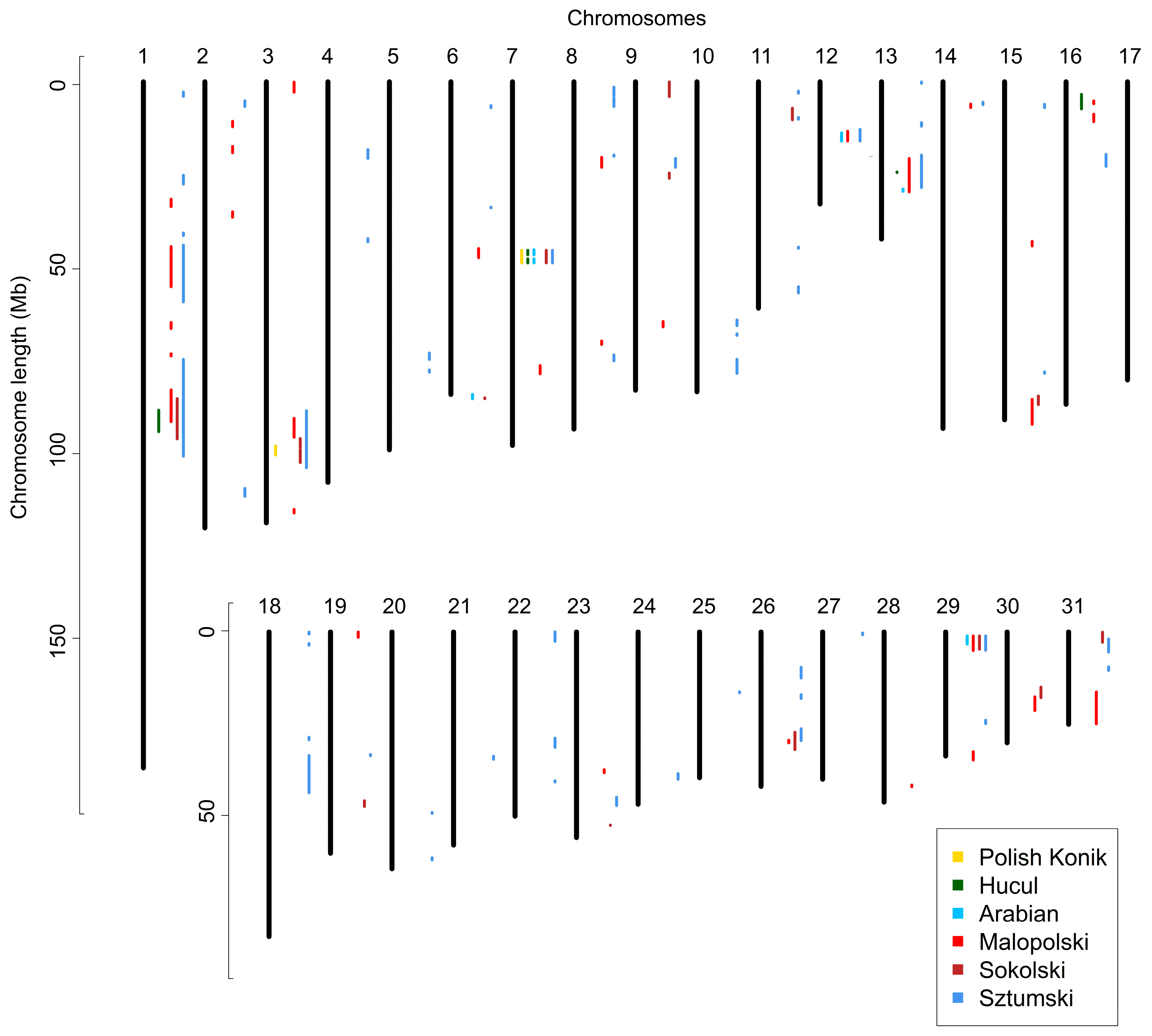
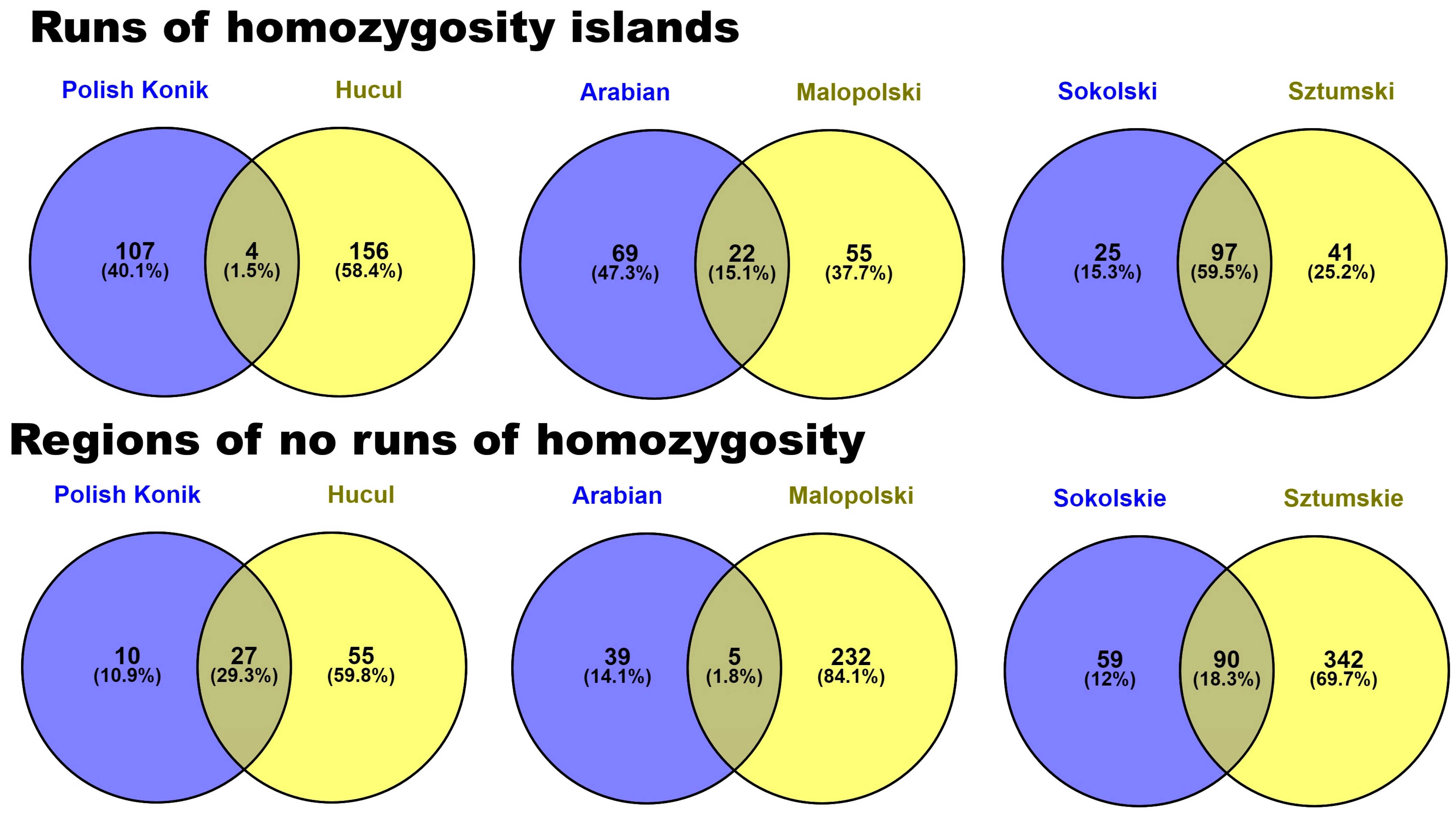
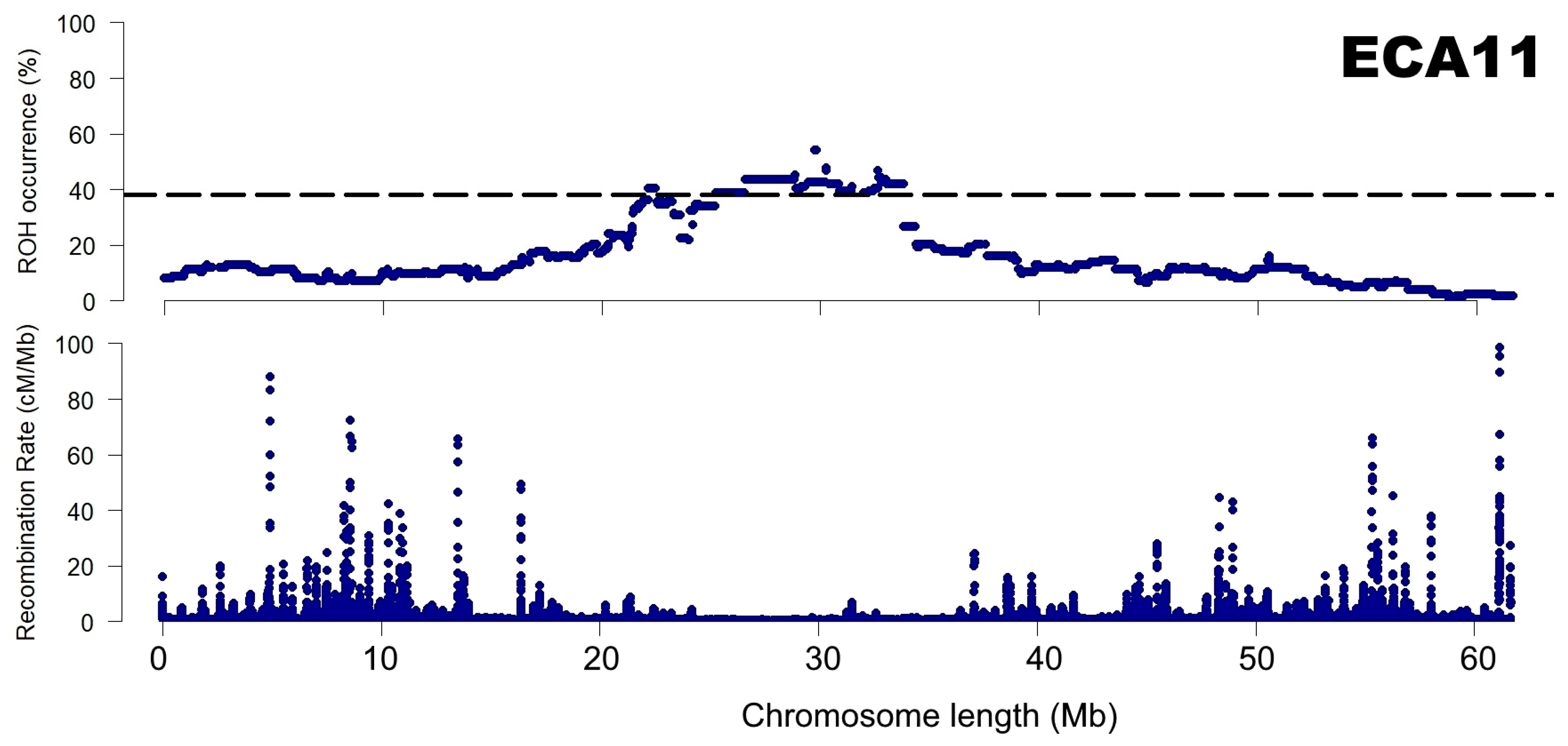
3.2. Identification of ROH Islands and No-ROH Regions
4. Discussion
4.1. ROH Characteristics
4.2. ROH Patterns
No ROH Regions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curik, I.; Ferencakovic, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Ferencakovic, M.; Hamzic, E.; Gredler, B.; Curik, I.; Sölkner, J. Runs of homozygosity reveal genomewide autozygosity in the Austrian Fleckvieh cattle. Agric. Conspec. Sci. 2011, 76, 325–328. [Google Scholar]
- Szmatola, T.; Gurgul, A.; Jasielczuk, I.; Zabek, T.; Ropka-Molik, K.; Litwińczuk, Z.; Bugno-Poniewierska, M. A Comprehensive Analysis of Runs of Homozygosity of Eleven Cattle Breeds Representing Different Production Types. Animals 2019, 9, 1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howrigan, D.P.; Simonson, M.A.; Keller, M.C. Detecting autozygosity through runs of homozygosity: A comparison of three autozygosity detection algorithms. BMC Genom. 2011, 12, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrangelo, S.; Tolone, M.; Gerlando, R.D.; Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothnagel, M.; Lu, T.T.; Kayser, M.; Krawczak, M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum. Mol. Genet. 2010, 19, 2927–2935. [Google Scholar] [CrossRef] [Green Version]
- Pemberton, T.J.; Absher, D.; Feldmand, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 2012, 10, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Solkner, J.; Ferencakovic, M.; Gredler, B.; Curik, I. Genomic metrics of individual autozygosity, applied to a cattle population, In Proceedings of the 61st Annual Meeting of the European Association of Animal Production, Heraklion, Greece, 22–26 August 2010.
- Ferencakovic, M.; Hamzic, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Solkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassell, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of artificial selection on runs of homozygosity in U. S. Holstein cattle, PLoS ONE 2013, 8, e80813. [Google Scholar]
- Librado, P.; Khan, N.; Fages, A.; Kusliy, M.A.; Suchan, T.; Tonasso-Calvière, L.; Schiavinato, S.; Alioglu, D.; Fromentier, A.; Perdereau, A.; et al. The origins and spread of domestic horses from the Western Eurasian steppes Pre-domestication population structure. Nature 2021, 598, 634–640. [Google Scholar] [CrossRef]
- Remer, V.; Bozlak, E.; Felkel, S.; Radovic, L.; Rigler, D.; Grilz-Seger, G.; Stefaniuk-Szmukier, M.; Bugno-Poniewierska, M.; Brooks, S.; Miller, D.C.; et al. Y-Chromosomal Insights into Breeding History and Sire Line Genealogies of Arabian Horses. Genes 2022, 13, 229. [Google Scholar] [CrossRef]
- Cosgrove, E.J.; Sadeghi, R.; Schlamp, F.; Holl, H.M.; Moradi-Shahrbabak, M.; Miraei-Ashtiani, S.R.; Abdalla, S.; Shykind, B.; Troedsson, M.; Stefaniuk-Szmukier, M.; et al. Genome Diversity and the Origin of the Arabian Horse. Sci. Rep. 2020, 10, 9702. [Google Scholar] [CrossRef]
- Dubois, C.; Ricard, A. Efficiency of past selection of the French Sport Horse: Selle Français breed and suggestions for the future. Livest. Sci. 2007, 112, 161–171. [Google Scholar] [CrossRef]
- Prawocheński, R. Hodowla Koni; PWRiL: Warszawa, Poland, 2010. [Google Scholar]
- Fornal, A.; Radko, A.; Nogaj, J.; Zielinska-Darecka, K.; Piestrzynska-Kajtoch, A. Malopolski Horse Stallions: Genetic Diversity Estimated on the Basisof Microsatellite DNA and Class I Markers. Folia Bilogica 2018, 66, 83–87. [Google Scholar] [CrossRef]
- Polak, G.; Gurgul, A.; Jasielczuk, I.; Szmatoła, T.; Edrzej Krupí Nski, J.; Bugno-Poniewierska, M. Suitability of Pedigree Information and Genomic Methods for Analyzing Inbreeding of Polish Cold-Blooded Horses Covered by Conservation Programs. Genes 2021, 12, 429. [Google Scholar] [CrossRef]
- Jasielczuk, I.; Gurgul, A.; Szmatoła, T.; Semik-Gurgul, E.; Pawlina-Tyszko, K.; Stefaniuk-Szmukier, M.; Polak, G.; Tomczyk-Wrona, I.; Bugno-Poniewierska, M. Linkage disequilibrium, haplotype blocks and historical effective population size in Arabian horses and selected Polish native horse breeds. Livest. Sci. 2020, 239, 104095. [Google Scholar] [CrossRef]
- Gurgul, A.; Jasielczuk, I.; Semik-Gurgul, E.; Pawlina-Tyszko, K.; Stefaniuk-Szmukier, M.; Szmatoła, T.; Polak, G.; Tomczyk-Wrona, I.; Bugno-Poniewierska, M. A genome-wide scan for diversifying selection signatures in selected horse breeds. PLoS ONE 2019, 14, e0210751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasicka. Polish Konik Horse–Characteristics and historical background of native descendants of tarpan. Acta Sci. Pol. 2013, 12, 25–38. [Google Scholar]
- Pasternak, M.; Krupinski, J.; Gurgul, A.; Bugno-Poniewierska, M. Genetic, historical and breeding aspects of the occurrence of the tobiano pattern and white markings in the Polish population of Hucul horses–a review. J. Appl. Anim. Res. 2020, 48, 21–27. [Google Scholar] [CrossRef]
- Gurgul, A.; Jasielczuk, I.; Semik-Gurgul, E.; Pawlina-Tyszko, K.; Szmatola, T.; Polak, G.; Bugno-Poniewierska, M. Genetic Differentiation of the Two Types of Polish Cold-blooded Horses Included in the National Conservation Program. Animals 2020, 10, 542. [Google Scholar] [CrossRef] [Green Version]
- Tomczyk-Wrona, I. Charakterystyka udziału ras tworzących w populacji ogierów małopolskich uznanych do programu ochrony zasobów genetycznych koni rasy małopolskiej. Wiad. Zoot. 2014, 4, 125–135. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, 2020, Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 10 November 2022).
- Beeson, S.K.; Mickelson, J.R.; McCue, M.E. Equine recombination map updated to EquCab3.0. Anim. Genet. 2019, 51, 341–342. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, 884–891. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Druml, T.; Neuditschko, M.; Mesaric, M.; Cotman, M.; Brem, G. Analysis of ROH patterns in the Noriker horse breed reveals signatures of selection for coat color and body size. Anim. Genet. 2019, 50, 334–346. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Mesaric, M.; Cotman, M.; Neuditschko, M.; Druml, T.; Brem, G. Runs of homozygosity and population history of three horse breeds with small population size. J. Equine Vet. Sci. 2018, 71, 27–34. [Google Scholar] [CrossRef]
- Druml, T.; Neuditschko, M.; Grilz-Seger, G.; Horna, M.; Ricard, A.; Mesaric, M.; Cotman, M.; Pausch, H.; Brem, G. Population Networks Associated with Runs of Homozygosity Reveal New Insights into the Breeding History of the Haflinger Horse. J. Hered. 2018, 109, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Gorssen, W.; Meyermans, R.; Janssens, S.; Buys, N. A publicly available repository of ROH islands reveals signatures of selection in different livestock and pet species. GSE 2021, 53, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; MacCiotta, N.P.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Fornal, A.; Kowalska, K.; Zabek, T.; Piestrzynska-Kajtoch, A.; Musiał, A.D.; Ropka-Molik, K. Genetic Diversity and Population Structure of Polish Konik Horse Based on Individuals from All the Male Founder Lines and Microsatellite Markers. Animals 2020, 10, 1569. [Google Scholar] [CrossRef]
- Kondás, K.; Szláma, G.; Trexler, M.; Patthy, L. Both WFIKKN1 and WFIKKN2 have high affinity for growth and differentiation factors 8 and 11. J. Biol. Chem. 2008, 283, 23677–23684. [Google Scholar] [CrossRef] [Green Version]
- Monestier, O.; Blanquet, V. WFIKKN1 and WFIKKN2: “Companion” proteins regulating TGFB activity. Cytokine Growth Factor Rev. 2016, 32, 75–84. [Google Scholar] [CrossRef]
- Haidet, A.M.; Rizo, L.; Handy, C.; Umapathi, R.; Eagle, A.; Shilling, C.; Boue, D.; Martin, P.T.; Sahenk, Z.; Mendell, J.R.; et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 4318–4322. [Google Scholar] [CrossRef] [Green Version]
- Brun, C.; Monestier, O.; Legardinier, S.; Maftah, A.; Blanquet, V. Murine GASP-1 N-Glycosylation is not essential for its activity on C2C12 myogenic cells but alters its secretion. Cell Physiol. Biochem. 2012, 30, 791–804. [Google Scholar] [CrossRef]
- Spurlin, B.A.; Park, S.Y.; Nevins, A.K.; Kim, J.K.; Thurmond, D.C. Syntaxin 4 transgenic mice exhibit enhanced insulin-mediated glucose uptake in skeletal muscle. Diabetes 2004, 53, 2223–2231. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.; Kim, B.G.; Lee, S.J.; Jeong, H.J.; Park, J.W.; Seo, D.W.; Kim, Y.K.; Lee, H.Y.; Han, J.W.; Kang, J.S.; et al. Syntaxin 4 regulates the surface localization of a promyogenic receptor Cdo thereby promoting myogenic differentiation. Skelet Muscle 2015, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, Y.; Calhabeu, F.; Morgan, J.E.; Katagiri, T.; Amthor, H.; Zammit, P.S. BMP signalling permits population expansion by preventing premature myogenic differentiation in muscle satellite cells. Cell Death Differ. 2011, 18, 222–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costamagna, D.; Mommaerts, H.; Sampaolesi, M.; Tylzanowski, P. Noggin inactivation affects the number and differentiation potential of muscle progenitor cells in vivo. Sci. Rep. 2016, 6, 31949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandini, M.A.; Felix, R. Molecular and functional interplay of voltage-gated Ca2⁺ channels with the cytoskeleton. Curr. Mol. Pharmacol. 2015, 8, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Guo, X.; Chen, S.-Y. Olfactomedin 2, a novel regulator for transforming growth factor-β-induced smooth muscle differentiation of human embryonic stem cell-derived mesenchymal cells. Mol. Biol. Cell 2014, 25, 4106–4114. [Google Scholar] [CrossRef] [PubMed]
- Shimoide, T.; Kawao, N.; Morita, H.; Ishida, M.; Takafuji, Y.; Kaji, H. Roles of Olfactomedin 1 in Muscle and Bone Alterations Induced by Gravity Change in Mice. Calcif. Tissue Int. 2020, 107, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Bryan, K.; McGivney, B.A.; Farries, G.; McGettigan, P.A.; McGivney, C.L.; Gough, K.F.; MacHugh, D.E.; Katz, L.M.; Hill, E.W. Equine skeletal muscle adaptations to exercise and training: Evidence of differential regulation of autophagosomal and mitochondrial components. BMC Genom. 2017, 18, 595. [Google Scholar] [CrossRef] [Green Version]
- Kruger, K.; Reichel, T.; Zeilinger, C. Role of heat shock proteins 70/90 in exercise physiology and exercise immunology and their diagnostic potential in sports. J. Physiol. 2019, 4, 916–927. [Google Scholar] [CrossRef]
- Denham, J.; McCluskey, M.; Denham, M.M.; Sellami, M.; Davie, A.J. Epigenetic control of exercise adaptations in the equine athlete: Current evidence and future directions. Equine Vet. J. 2021, 53, 431–450. [Google Scholar] [CrossRef]
- Voisin, S.; Eynon, N.; Yan, X.; Bishop, D.J. Exercise training and DNA methylation in humans. Acta Physiol. 2015, 213, 39–59. [Google Scholar] [CrossRef]
- Zhao, W.; Mu, Y.; Ma, L.; Wang, C.; Tang, Z.; Yang, S.; Zhou, R.; Hu, X.; Li, M.H.; Li, K. Systematic identification and characterization of long intergenic non-coding RNAs in fetal porcine skeletal muscle development. Sci. Rep. 2015, 5, 8957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, T.C.; Morris, K.V.; Weinberg, M.S. Perspectives on the mechanism of transcriptional regulation by long non-coding RNAs. Epigenetics 2014, 9, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Bonilauri, B.; Dallagiovanna, B. Long Non-coding RNAs Are Differentially Expressed After Different Exercise Training Programs. Front. Physiol. 2020, 11, 567614. [Google Scholar] [CrossRef]
- Zheng, L.; Liu, X.; Chen, P.; Xiao, W. Expression and role of lncRNAs in the regeneration of skeletal muscle following contusion injury. Exp. Ther. Med. 2019, 18, 2617–2627. [Google Scholar] [CrossRef] [PubMed]
- Emerson, R.O.; Thomas, J.H. Adaptive Evolution in Zinc Finger Transcription Factors. PLoS Genet. 2009, 5, e1000325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nail, A.N.; Smith, J.J.; Peterson, M.L.; Spear, B.T. Evolutionary Analysis of the Zinc Finger and Homeoboxes Family of Proteins Identifies Multiple Conserved Domains and a Common Early Chordate Ancestor. Genome Biol. Evol. 2020, 12, 174–184. [Google Scholar] [CrossRef]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschellà, G. Zinc-finger proteins in health and disease. Cell Death Discov. 2017, 3, 17071. [Google Scholar] [CrossRef] [Green Version]
- Sevane, N.; Dunner, S.; Boado, A.; Cañon, J. Polymorphisms in ten candidate genes are associated with conformational and locomotive traits in Spanish Purebred horses. J. Appl. Genet. 2016, 58, 355–361. [Google Scholar] [CrossRef]
- Boyko, A.R.; Brooks, S.A.; Behan-Braman, A.; Castelhano, M.; Corey, E.; Oliveira, K.C.; Swinburne, J.E.; Todhunter, R.J.; Zhang, Z.; Ainsworth, D.M.; et al. Genomic analysis establishes correlation between growth and laryngeal neuropathy in Thoroughbreds. BMC Genom. 2014, 15, 259. [Google Scholar] [CrossRef] [Green Version]
- Metzger, J.; Schrimpf, R.; Philipp, U.; Distl, O. Espression levels of LCORL are associated with body size in horses. PLoS ONE 2013, 8, e56497. [Google Scholar] [CrossRef] [Green Version]
- Mackiewicz, D.; Oliveira, P.M.C.; Oliveira, S.M.; Cebrat, S. Distribution of Recombination Hotspots in the Human Genome–A Comparison of Computer Simulations with Real Data. PLoS ONE 2013, 8, e65272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisen, D.P.; Osthoff, M. If there is an evolutionary selection pressure for the high frequency of MBL2 polymorphisms, what is it? Clin. Exp. Immunol. 2014, 176, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Fijarczyk, A.; Babik, W. Detecting balancing selection in genomes: Limits and Prospects. Mol. Ecol. 2015, 24, 3529–3545. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, G.; Ruan, D.; Zhuang, Z.; Ding, R.; Quan, J.; Wang, S.; Jiang, Y.; Huang, J.; Gu, T.; et al. Runs of Homozygosity Uncover Potential Functional-Altering Mutation Associated With Body Weight and Length in Two Duroc Pig Lines. Front. Vet. Sci. 2022, 9, 832633. [Google Scholar] [CrossRef]
- Ke, T.; Gomez, C.R.; Mateus, H.E.; Castano, J.A.; Wang, Q.K. Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a South American family. J. Hum. Genet. 2009, 54, 660–664. [Google Scholar] [CrossRef] [Green Version]
- Sintas, C.; Carreño, O.; Fernàndez-Castillo, N.; Corominas, R.; Vila-Pueyo, M.; Toma, C.; Cuenca-León, E.; Barroeta, I.; Roig, C.; Volpini, V.; et al. Mutation Spectrum in the CACNA1A Gene in 49 Patients with Episodic Ataxia. Sci. Rep. 2017, 7, 2514. [Google Scholar] [CrossRef]
- Algahtani, H.; Shirah, B.; Algahtani, R.; Al-Qahtani, M.H.; Abdulkareem, A.A.; Naseer, M.I. A novel mutation in CACNA1A gene in a Saudi female with episodic ataxia type 2 with no response to acetazolamide or 4-aminopyridine. Intractable Rare Dis. Res. 2019, 8, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Ablondi, M.; Viklund, A.; Lindgren, G.; Eriksson, S.; Mikko, S. Signatures of selection in the genome of Swedish warmblood horses selected for sport performance. BMC Genom. 2019, 20, 717. [Google Scholar] [CrossRef]
- Macario, A.J.L.; Grippo, T.M.; Macario, E.C. Genetic disorders involving molecular-chaperone genes: A perspective. Genet. Med. 2005, 7, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Torres, F.; Aranega, A.E.; Franco, D. Identification of regulatory elements directing miR-23a-miR-27a-miR-24-2 transcriptional regulation in response to muscle hypertrophic stimuli. Biochim. Biophys. Acta 2014, 1839, 885–897. [Google Scholar] [CrossRef]
- Chinchilla, A.; Lozano, E.; Daimi, H.; Esteban, F.J.; Crist, C.; Aranega, A.E.; Franco, D. MicroRNA profiling during mouse ventricular maturation: A role for miR-27 modulating Mef2c expression. Cardiovasc. Res. 2011, 89, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano-Velasco, E.; Contreras, A.; Crist, C.; Hernández-Torres, F.; Franco, D.; Aránega, A.E. Pitx2c modulates Pax3 +/Pax7 + cell populations and regulates Pax3 expression by repressing miR27 expression during myogenesis. Dev. Biol. 2011, 357, 165–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Zhang, Y.; Yang, G.; Chen, X.; Zhang, Y.; Cao, G.; Wang, J.; Sun, Y.; Zhang, P.; Fan, M.; et al. Transforming growth factor-β-regulated miR-24 promotes skeletal muscle differentiation. Nucleic Acids. Res. 2008, 36, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Hudson, M.B.; Woodworth-Hobbs, M.E.; Zheng, B.; Rahnert, J.A.; Blount, M.A.; Gooch, J.L.; Searles, C.D.; Price, S.R. miR-23a is decreased during muscle atrophy by a mechanism that includes calcineurin signaling and exosome-mediated export. Am. J. Physiol. Cell Physiol. 2014, 306, C551–C558. [Google Scholar] [CrossRef]
- Okamura, T.; Hashimoto, Y.; Osaka, T.; Senmaru, T.; Fukuda, T.; Hamaguchi, M.; Fukui, M. miR-23b-3p acts as a counter-response against skeletal muscle atrophy. J. Endocrinol. 2020, 244, 535–547. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, Q.; Sun, J.; Li, X.; Xu, L.; Yang, H.; Shi, H.; Ning, S.; Chen, L.; Li, Y.; et al. In silico detection and characteristics of novel microRNA genes in the Equus caballus genome using an integrated ab initio and comparative genomic approach. Genomics 2009, 94, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Ferenčaković, M.; Solkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Number of Individuals | Females | Males | Horse Type |
|---|---|---|---|---|
| Polish Konik | 99 | 71 | 28 | Primitive |
| Hucul | 116 | 77 | 39 | Primitive |
| Arabian | 124 | 91 | 33 | Light |
| Malopolski | 56 | 50 | 6 | Light |
| Sokolski | 107 | 86 | 21 | Draft |
| Sztumski | 69 | 69 | 0 | Draft |
| Breeds | Number of Genes | Gene Names |
|---|---|---|
| AR HC KP SOK SZTUM | 27 | PODNL1 KLF1 TRMT1 DNAJB1 NDUFB7 DNASE2 ADGRE3 eca-mir-8997 CACNA1A PALM3 ASF1B GADD45GIP1 FARSA eca-mir-24-1 LYL1 PTGER1 STX10 ADGRL1 DDX39A RTBDN RFX1 BRME1 C7H19orf67 PRKACA GIPC1 eca-mir-23a eca-mir-1271b |
| HC MLP SOK SZTUM | 9 | MEX3B EFL1 STARD5 U6 TMC3 MAT1A DYDC1 MESD SFTPA1 |
| MLP SOK SZTUM | 5 | LRIT1 FZD8 LRIT2 CUL2 PARD3 |
| HC SOK SZTUM | 25 | NMB KIF7 FSD2 RCCD1 AP3S2 HOMER2 MAN2A2 IQGAP1 BTBD1 IDH2 MESP1 FES SEC11A CRTC3 FURIN CIB1 HDGFL3 BLM PEX11A eca-mir-9055 UNC45A AP3B2 WDR73 BNC1 PLIN1 |
| KP SOK SZTUM | 9 | SEL1L3 DHPS TRIR HOOK2 CCKAR PRDX2 FBXW9 WDR83OS TNPO2 |
| AR MLP SZTUM | 1 | OR4C5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szmatoła, T.; Gurgul, A.; Jasielczuk, I.; Oclon, E.; Ropka-Molik, K.; Stefaniuk-Szmukier, M.; Polak, G.; Tomczyk-Wrona, I.; Bugno-Poniewierska, M. Assessment and Distribution of Runs of Homozygosity in Horse Breeds Representing Different Utility Types. Animals 2022, 12, 3293. https://doi.org/10.3390/ani12233293
Szmatoła T, Gurgul A, Jasielczuk I, Oclon E, Ropka-Molik K, Stefaniuk-Szmukier M, Polak G, Tomczyk-Wrona I, Bugno-Poniewierska M. Assessment and Distribution of Runs of Homozygosity in Horse Breeds Representing Different Utility Types. Animals. 2022; 12(23):3293. https://doi.org/10.3390/ani12233293
Chicago/Turabian StyleSzmatoła, Tomasz, Artur Gurgul, Igor Jasielczuk, Ewa Oclon, Katarzyna Ropka-Molik, Monika Stefaniuk-Szmukier, Grazyna Polak, Iwona Tomczyk-Wrona, and Monika Bugno-Poniewierska. 2022. "Assessment and Distribution of Runs of Homozygosity in Horse Breeds Representing Different Utility Types" Animals 12, no. 23: 3293. https://doi.org/10.3390/ani12233293
APA StyleSzmatoła, T., Gurgul, A., Jasielczuk, I., Oclon, E., Ropka-Molik, K., Stefaniuk-Szmukier, M., Polak, G., Tomczyk-Wrona, I., & Bugno-Poniewierska, M. (2022). Assessment and Distribution of Runs of Homozygosity in Horse Breeds Representing Different Utility Types. Animals, 12(23), 3293. https://doi.org/10.3390/ani12233293