
Genomic Prediction Accuracies for Growth and Carcass Traits in a Brangus Heifer Population

 , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotypes
2.2. SNP Marker Genotypes
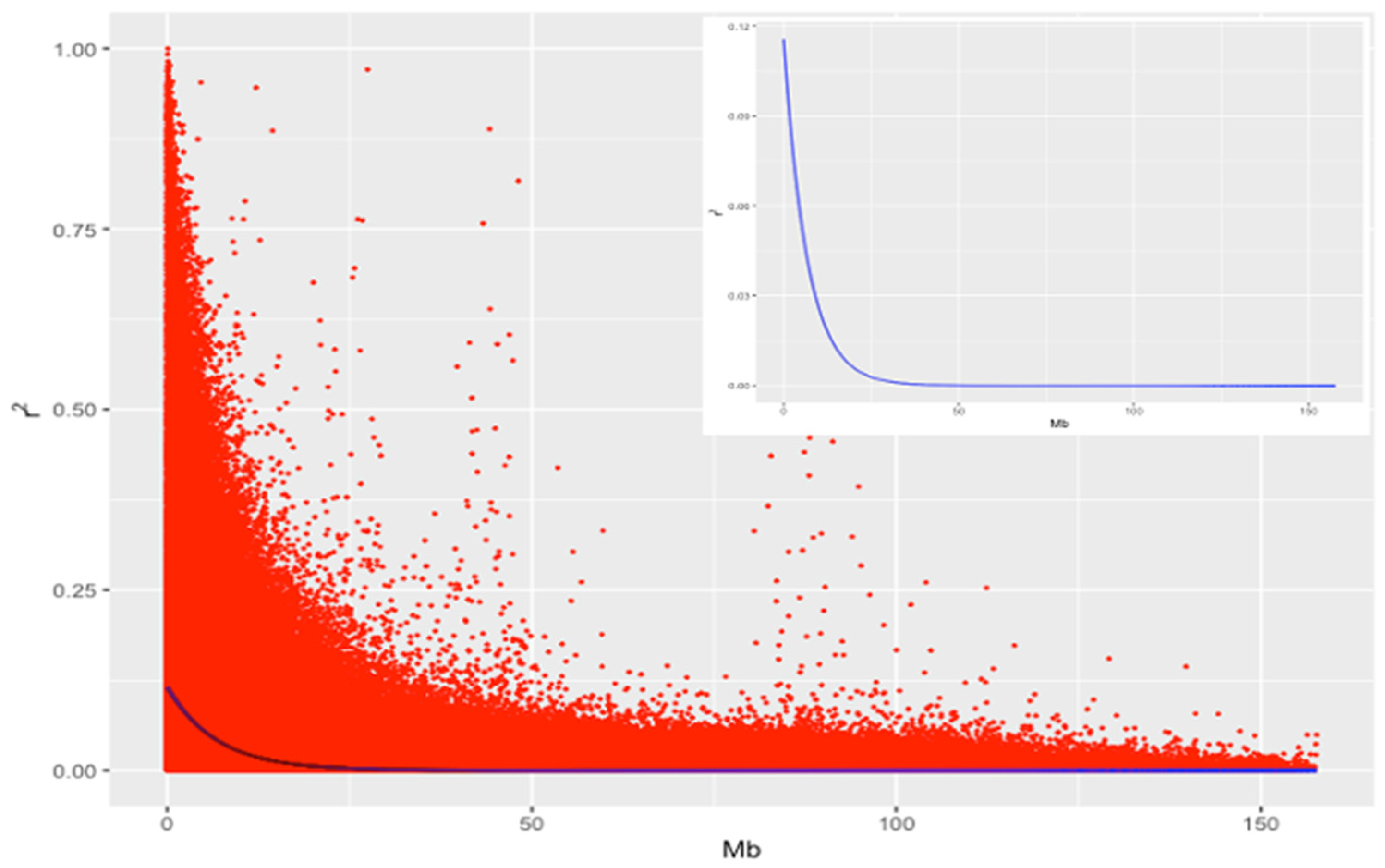
2.3. Linkage Disequilibrium
2.4. Genomic Selection
2.4.1. Genomic Best Linear Unbiased Prediction (GBLUP)
2.4.2. The Bayesian BayesA, BayesB, BayesC and Lasso Methods
2.4.3. K-Means and Random Clustering
2.4.4. Accuracy of Genomic Prediction
3. Results and Discussion
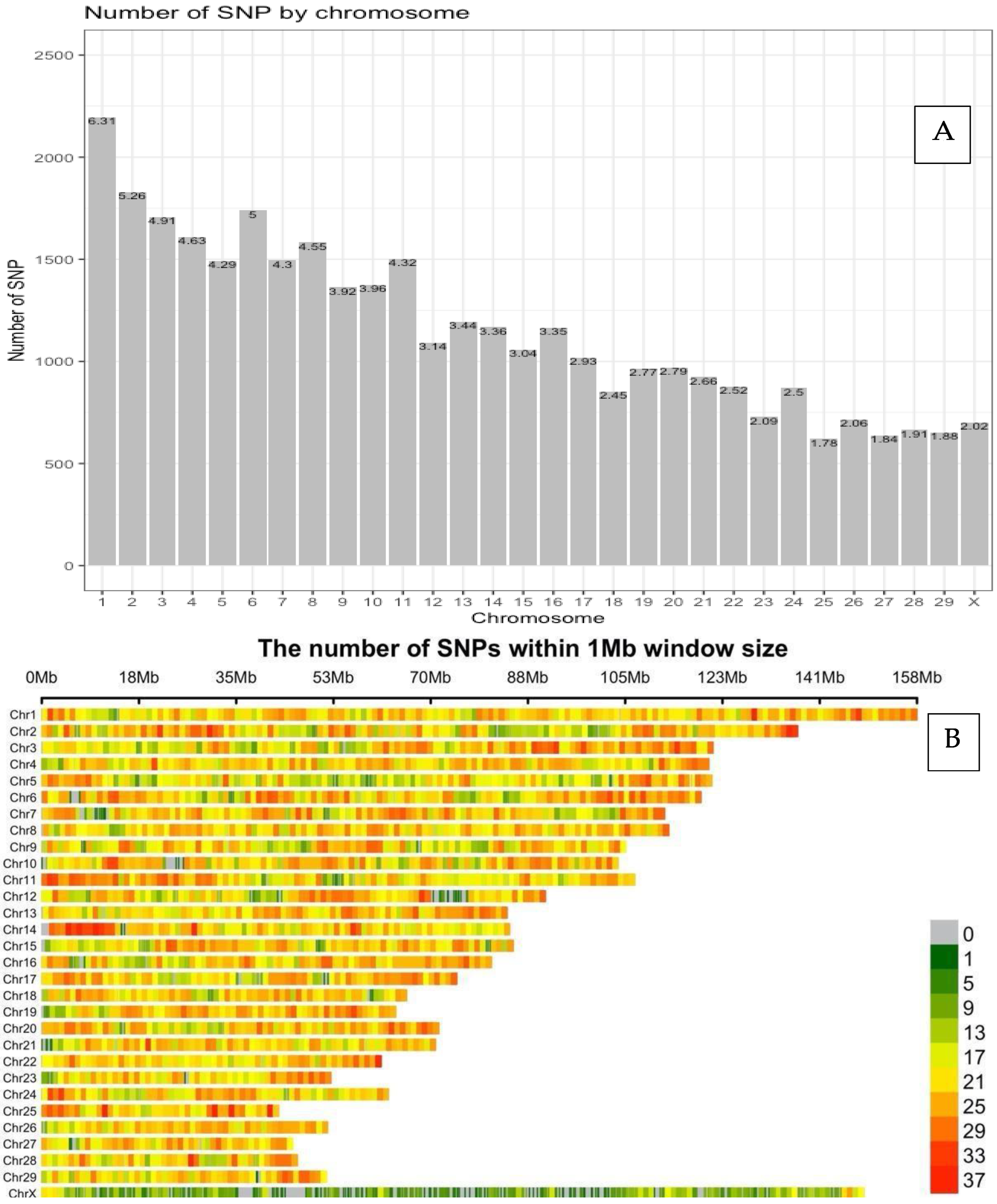
3.1. Distribution of SNP Markers and LD Analysis
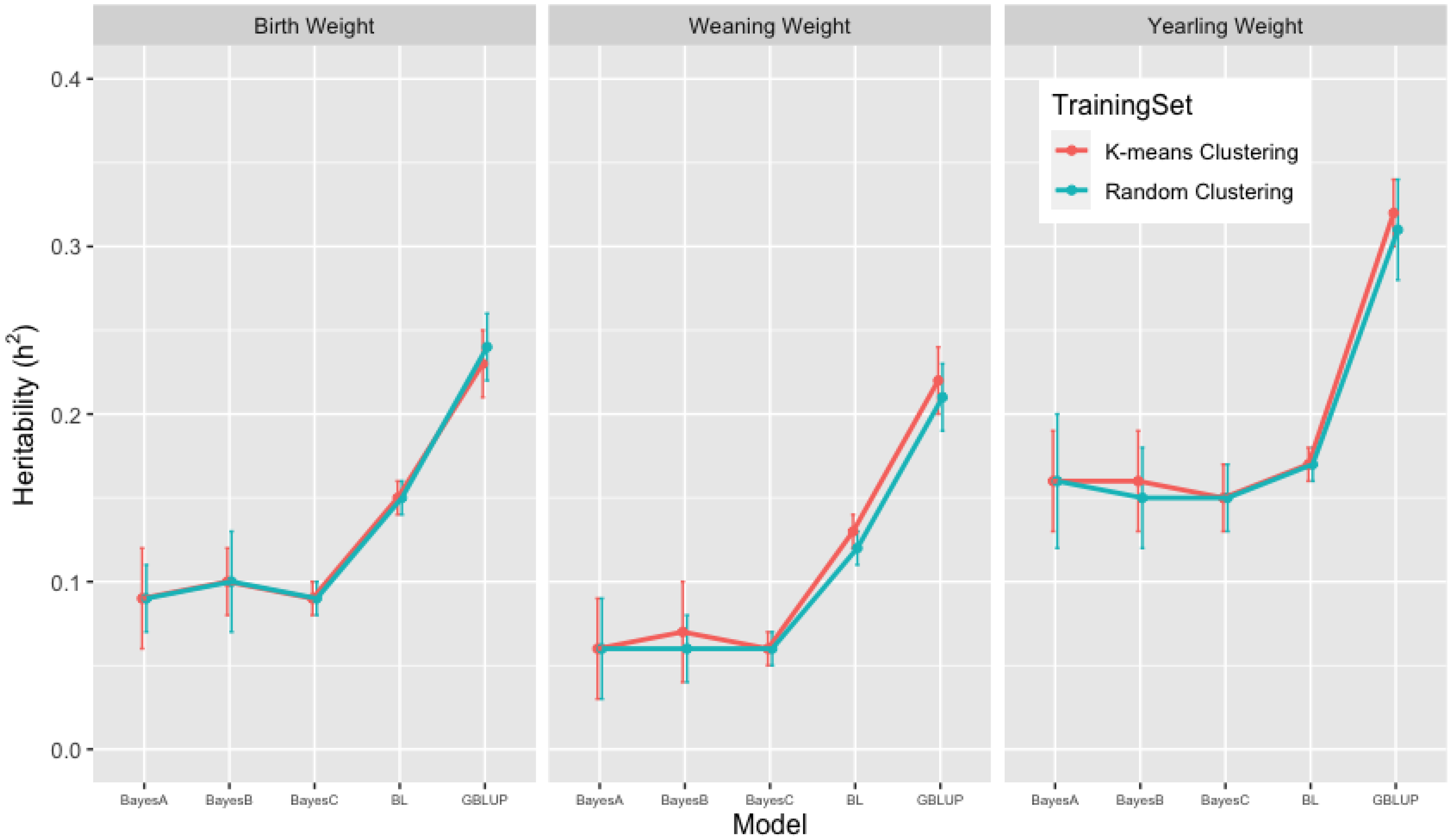
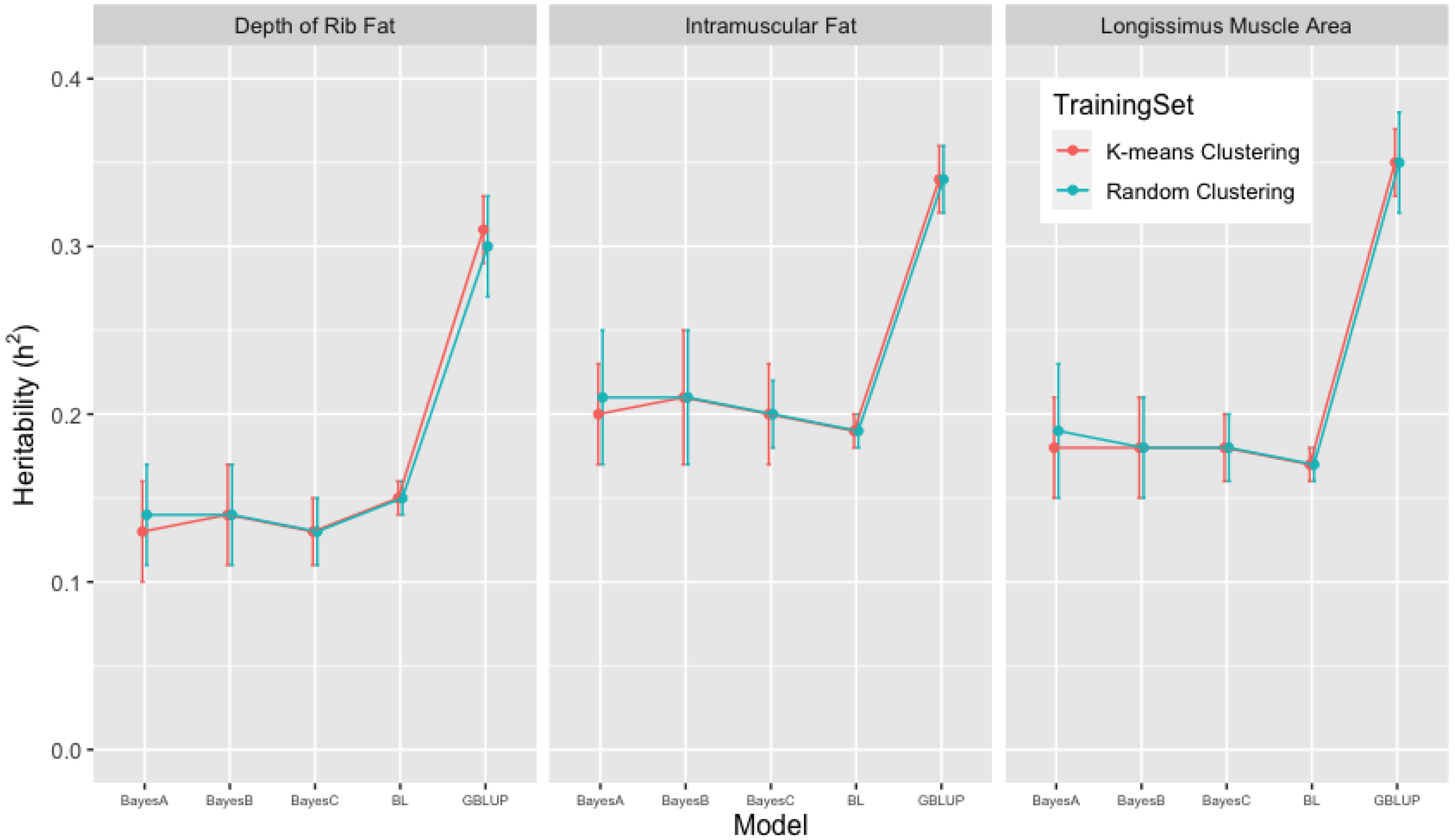
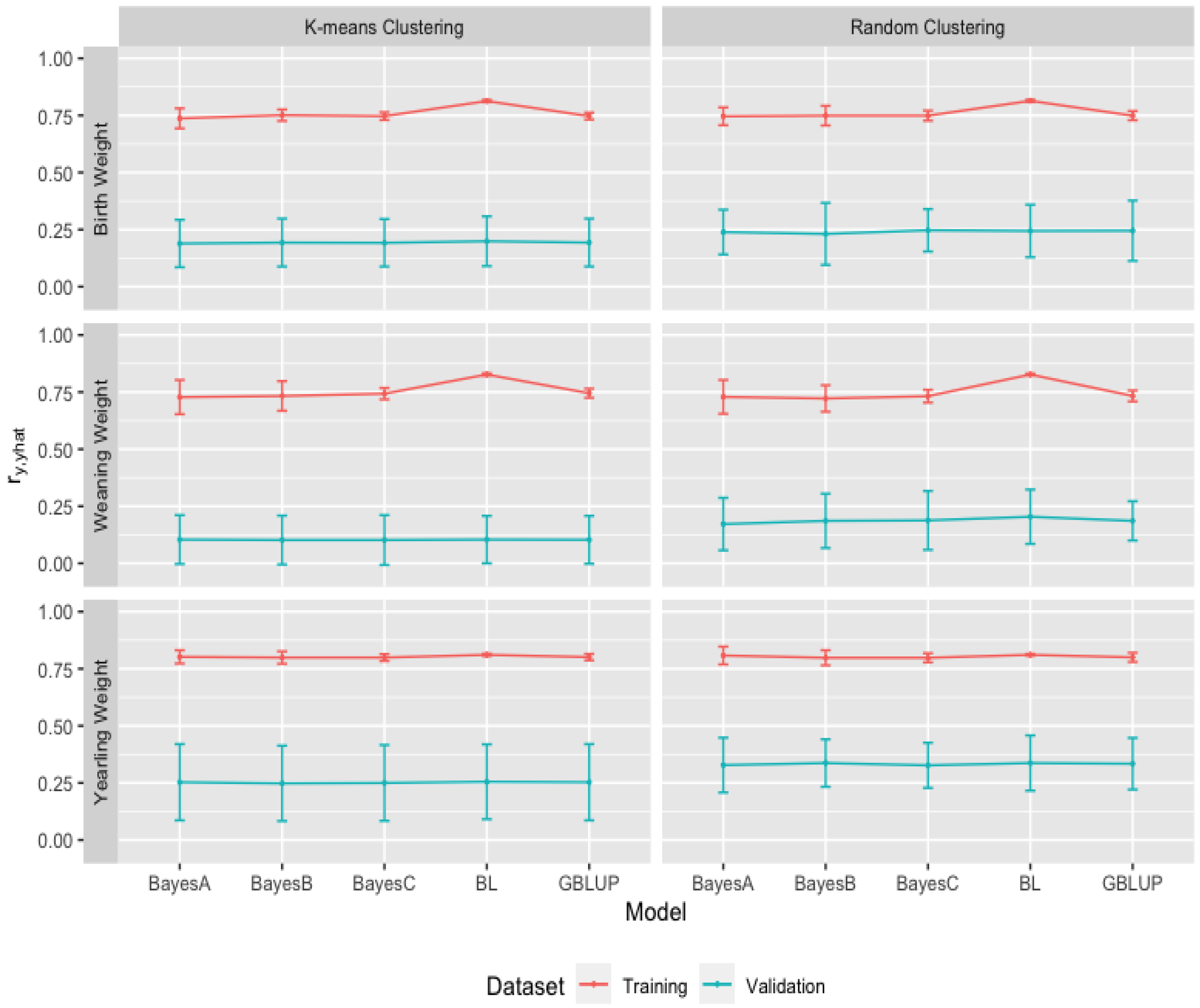
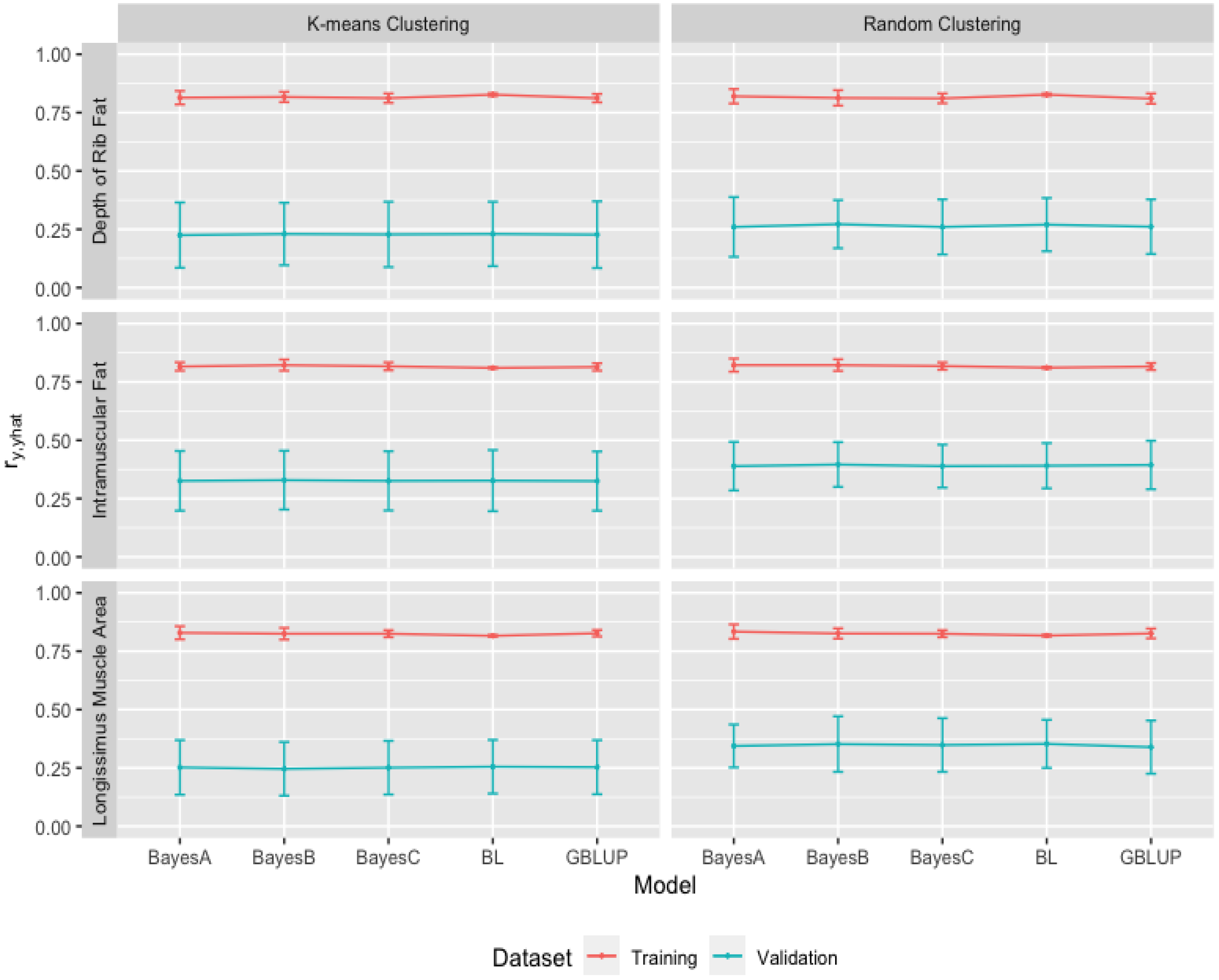
3.2. Heritability Estimates from GBLUP, and the Bayesian (BayesA, BayesB, BayesC and Lasso) Methods in K-Means and Random Training Datasets
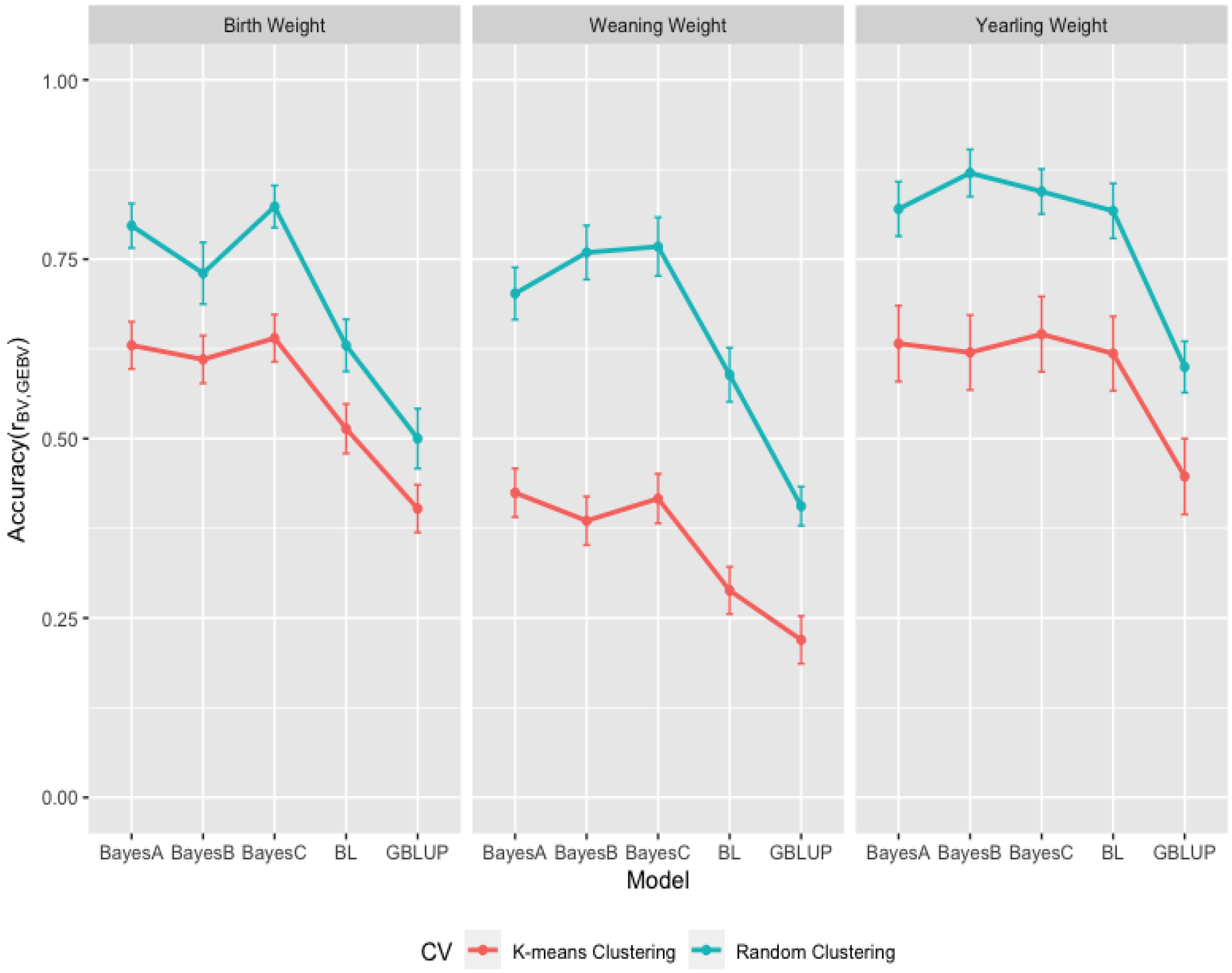
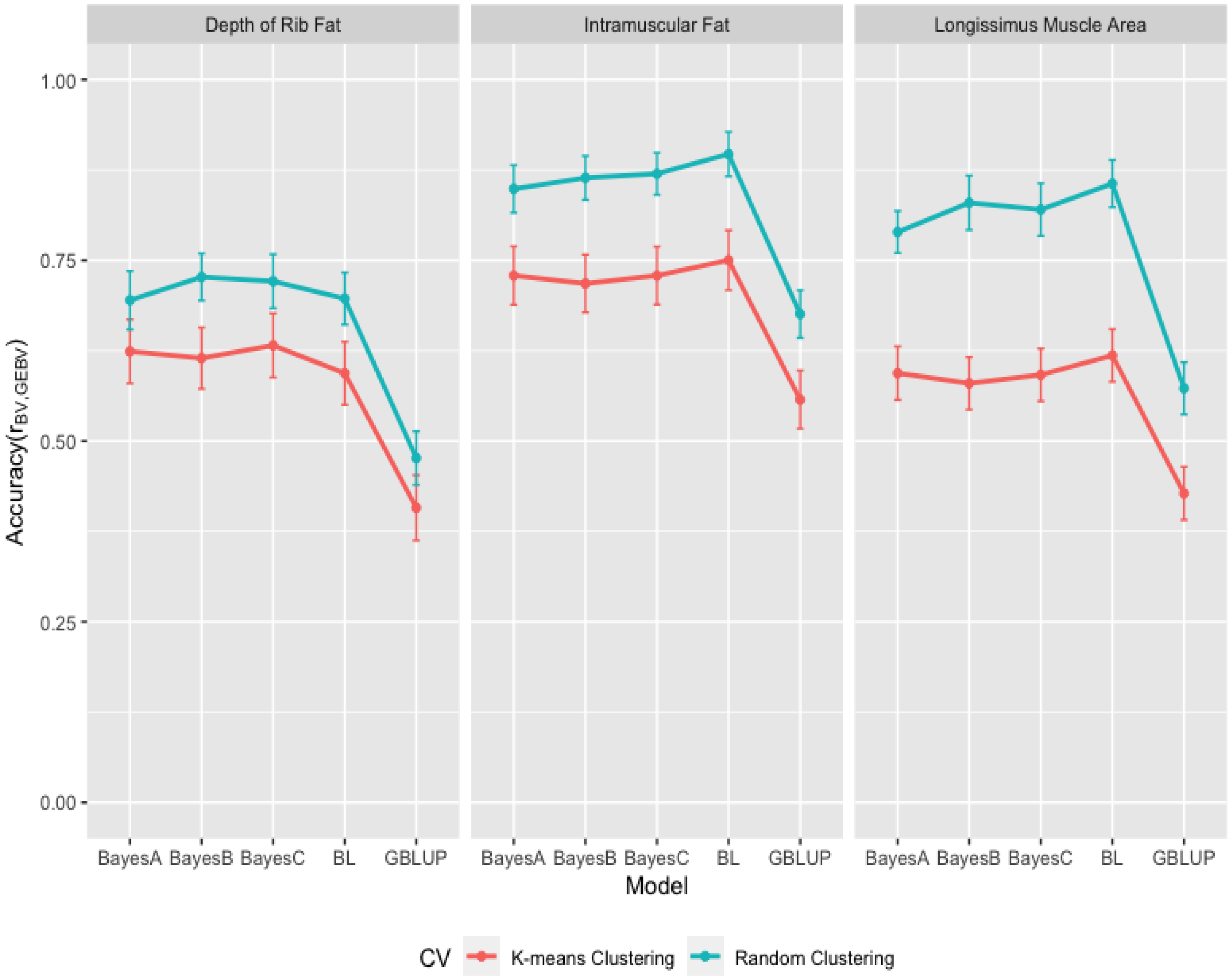
3.3. Comparison of Genome-Wide Prediction Ability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.L.; Sonstegard, T.S.; et al. Development and characterization of high density SNP genotyping assay for cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef] [Green Version]
- Applied Biosystems. Axiom Bovine Genotyping v3 Array (384HT format). 2019. Available online: https://www.thermofisher.com/order/catalog/product/55108%209#/551089 (accessed on 15 January 2022).
- Illumina. Infinium iSelect Custom Genotyping Assays. 2016. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/technote_iselect_design.pdf (accessed on 15 January 2022).
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef] [Green Version]
- Pérez, P.; de los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.; Bowman, P.; Chamberlain, A.; Goddard, M. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanRaden, P.M.; Van Tassell, C.P.; Wiggans, G.; Sonstegard, T.; Schnabel, R.; Taylor, J.F.; Schenkel, F.S. Invited review: Reliability of genomic predictions for north American Holstein bulls. J. Dairy Sci. 2009, 92, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.O.; Kizilkaya, K.; Garrick, D.J.; Fernando, R.L.; Reecy, J.M.; Weaber, R.L.; Silver, G.A.; Thomas, M.G. Bayesian genome-wide association analysis of growth and yearling ultrasound measures of carcass traits in Brangus heifers. J. Anim. Sci. 2012, 90, 3398–3409. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.O.; Kizilkaya, K.; Garrick, D.J.; Fernando, R.L.; Reecy, J.M.; Weaber, R.L.; Silver, G.A.; Thomas, M.G. Heritability and Bayesian genome-wide association of binary traits of first service conception and heifer pregnancy in Brangus heifers. J. Anim. Sci. 2013, 91, 605–612. [Google Scholar] [CrossRef]
- de Los Campos, G.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P.L. Whole genome regression and prediction methods applied to plant and animal breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, T.; Hayes, B.; Goddard, M.E. Accelerating improvement of livestock with genomic selection. Annu. Rev. Anim. Biosci. 2013, 1, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.; Hayes, B.; Goddard, M.E. Genomic selec-tion: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Crossa, J.; de Los Campos, G.; Pérez, P.; Gianola, D.; Burgueño, J.; Araus, J.L.; Makumbi, D.; Singh, R.P.; Dreisigacker, S.; Yan, J.; et al. Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics 2010, 186, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombani, C.; Legarra, A.; Fritz, S.; Guillaume, F.; Croiseau, P.; Ducrocq, V.; Robert-Granié, C. Application of bayesian least absolute shrinkage and selection operator (LASSO) and BayesCπ methods for genomic selection in French holstein and montbéliarde breeds. J. Dairy Sci. 2013, 96, 575–591. [Google Scholar] [CrossRef] [Green Version]
- Esfandyari, H.; Sørensen, A.; Bijma, P.A. Crossbred reference population can improve the response to genomic selection for crossbred performance. Genet. Sel. Evol. 2015, 47, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhou, H.; Wu, Y.; Li, X.; Zhao, J.; Zuo, T.; Zhang, X.; Zhang, Y.; Liu, S.; Shen, Y.; et al. The impact of genetic relationship and linkage disequilibrium on genomic selection. PLoS ONE 2015, 10, e0132379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habier, D.; Fernando, R.L.; Dekkers, J.C.M. The impact of genetic relationship information of genome-assisted breeding values. Genetics 2007, 177, 2389–2397. [Google Scholar] [CrossRef] [Green Version]
- Saatchi, M.; McClure, M.C.; McKay, S.D.; Rolf, M.M.; Kim, J.; Decker, J.E.; Taxis, T.M.; Chapple, R.H.; Ramey, H.R.; Northcutt, S.L.; et al. Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet. Sel. Evol. 2011, 43, 40. [Google Scholar] [CrossRef] [Green Version]
- Villumsen, T.; Janss, L.; Lund, M. The importance of haplotype length and heritability using genomic selection in dairy cattle. J. Anim. Breed Genet Z. Tierz. Zucht. 2009, 126, 3–13. [Google Scholar] [CrossRef]
- Clark, S.; Hickey, J.; van der Werf, J. Different models of genetic variation and their effect on genomic evaluation. Genet Sel. Evol. 2011, 43, 18. [Google Scholar] [CrossRef] [Green Version]
- Luna-Nevarez, P.; Bailey, D.W.; Bailey, C.C.; VanLeeuwen, D.M.; Enns, R.M.; Silver, G.A.; DeAtley, K.L.; Thomas, M.G. Growth characteristics, reproductive performance, and evaluation of their associative relationships in Brangus cattle managed in a Chihuahuan Desert production system. J. Anim. Sci. 2010, 88, 1891–1904. [Google Scholar] [CrossRef] [Green Version]
- Fortes, M.R.S.; Snelling, W.M.; Reverter, A.; Nagaraji, S.H.; Lehnert, S.A.; Hawken, R.J.; DeAtley, K.L.; Peters, S.O.; Silver, G.A.; Rincon, G.; et al. Gene network analyses of first service conception in Brangus heifers: Use of genome and trait associations, hypo- thalamic-transcriptome information, and transcription factors. J. Anim. Sci. 2012, 90, 2894–2906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granato, I.S.C.; Galli, G.; de Oliveira Couto, E.G.; e Souza, M.B.; Mendonça, L.F.; Fritsche-Neto, R. snpReady: A tool to assist breeders in genomic analysis. Mol. Breeding 2018, 38, 102. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated tool for Genome-Wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 10 March 2022).
- Wimmer, V.; Albrecht, T.; Auinger, H.J.; Schon, C.C. Synbreed: A framework for the analysis of genomic prediction data using R. Bioinformatics 2012, 28, 2086–2087. [Google Scholar] [CrossRef] [Green Version]
- vanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Park, T.; Casella, G. The bayesian lasso. J. Am. Stat. Assoc. 2008, 103, 681–686. [Google Scholar] [CrossRef]
- Henderson, C.R. A simple method for computing the inverse of a numerator relationship matrix used in prediction of breeding values. Biometrics 1976, 32, 69. [Google Scholar] [CrossRef]
- Gorjanc, G.; Henderson, D.A.; Kinghorn, B.; Percy, A. GeneticsPed: Pedigree and Genetic Relationship Functions. 2020. R Package Version 1.52.0. Available online: http://rgenetics.org (accessed on 10 March 2022).
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. 2017. R Package Version 1.0.5. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 10 March 2022).
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A k-means clustering algorithm. Appl. Stat. 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Legarra, A.; Robert-Granié, C.; Manfredi, E.; Elsen, J.M. Performance of genomic selection in mice. Genetics 2008, 180, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, H.; Ahmad, N.; Heather, H.J.; Eui-soo, K.; Khan, W.A.; Ajmal, A.; Javed, K.; Pasha, T.N.; Ali, A.; Kim, J.J.; et al. Whole genome study of linkage disequilibrium in Sahiwal cattle. S. Afr. J. Anim. Sci. 2018, 48, 353–360. [Google Scholar] [CrossRef] [Green Version]
- El Hou, A.; Rocha, D.; Venot, E. Long-range linkage disequilibrium in French beef cattle breeds. Genet. Sel. Evol. 2021, 53, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kumar, A.; Mehrotra, A.; Pandey, A.K.; Mishra, B.P.; Dutt, T. Estimation of linkage disequilibrium levels and allele frequency distribution in crossbred Vrindavani cattle using 50K SNP data. PLoS ONE 2021, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Sargolzaei, M.; Kelly, M.; Li, C.; Vander Voort, G.; Wang, Z.; Plastow, G.; Moore, S.; Miller, S.P. Linkage disequilibrium in Angus, Charolais, and Crossbred beef cattle. Front. Gene. 2012, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- McKay, S.D.; Schnabel, R.D.; Murdoch, B.M.; Matukumalli, L.K.; Aerts, J.; Coppieters, W.; Crews, D.; Neto, E.D.; Gill, C.A.; Gao, C.; et al. Whole genome linkage disequilibrium maps in cattle. BMC Genet. 2007, 8, 74. [Google Scholar] [CrossRef] [Green Version]
- Ríos-Utrera, Á.; Vega-Murillo, V.E.; Martínez-Velázquez, G.; Montaño-Bermúdez, M. Comparison of models for the estimation of variance components for growth traits of registered limousin cattle. Trop. Subtrop. Agroecosyt. 2011, 14, 667–674. [Google Scholar]
- Neser, F.W.C.; van Wyk, J.B.; Fair, M.D.; Lubout, P.; Crook, B.J. Estimation of genetic parameters for growth traits in Brangus cattle. S. Afr. J. Anim. Sci. 2012, 42, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Rolf, M.M.; Garrick, D.J.; Fountain, T.; Ramey, H.R.; Weaber, R.L.; Decker, J.E.; Pollak, E.J.; Schnabel, R.D.; Taylor, J.F. Comparison of Bayesian models to estimate direct genomic values in multi-breed commercial beef cattle. Genet. Sel. Evol. 2015, 47, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, B.C.; Tholon, P.; Buzanskas, M.E.; Sbardella, A.P.; Rosa, J.O.; Campos da Silva, L.O.; Torres Júnior, R.A.A.; Munari, D.P.; Alencar, M.M. Genetic analyses on bodyweight, reproductive, and carcass traits in composite beef cattle. Anim. Prod. Sci. 2017, 57, 415–421. [Google Scholar] [CrossRef]
- Boldt, R.J. Genetic Parameters for Fertility and Production Traits in Red Angus Cattle; Master of Science, Colorado State University: Fort Collins, CO, USA, 2017. [Google Scholar]
- Habier, D.; Tetens, J.; Seefried, F.R.; Lichtner, P.; Thaller, G. The impact of genetic relationship information on genomic breeding values in German Holstein cattle. Genet. Sel. Evol. 2010, 42, 5. [Google Scholar] [CrossRef] [Green Version]
- Daetwyler, H.D.; Kemper, K.E.; Jh, V.D.W.; Hayes, B.J. Components of the accuracy of genomic prediction in a multi-breed sheep population. J. Anim. Sci. 2012, 90, 3375–3384. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, C.; Sargolzaei, M.; Schenkel, F. Impact of genotype imputation on the performance of GBLUP and Bayesian methods for genomic prediction. PLoS ONE 2014, 9, e101544. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Zhou, L.; Mrode, R.; Zhang, Q.; Liu, J.F. Incorporating single-step strategy into random regression model to enhance genomic prediction of longitudinal trait. Heredity 2016, 119, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Mrode, R.; Zhang, S.; Zhang, Q.; Li, B.; Liu, J. Factors affecting GEBV accuracy with single-step Bayesian models. Heredity 2018, 120, 100–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Habier, D.; Fernando, R.L.; Garrick, D.J.; Dekkers, J.C. Genomic breeding value prediction and QTL mapping of QTLMAS2010 data using Bayesian Methods. BMC Proc. 2011, 5, S13. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Su, G.; Janss, L.; Zhang, Y.; Lund, M. Model comparison on genomic predictions using high-density markers for different groups of bulls in the Nordic Holstein population. J. Dairy Sci. 2013, 96, 4678–4687. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Vinsky, M.; Li, C. Accuracy of predicting genomic breeding values for carcass merit traits in Angus and Charolais beef cattle. Anim. Genet. 2014, 46, 55–59. [Google Scholar] [CrossRef]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.C.; Verbyla, K.; Goddard, M.E. Accuracy of genomic breeding values in multi-breed dairy cattle populations. Genet. Sel. Evol. 2009, 41, 51. [Google Scholar] [CrossRef] [Green Version]
- Ostersen, T.; Christensen, O.F.; Henryon, M.; Nielsen, B.; Su, G.; Madsen, P. Deregressed EBV as the response variable yield more reliable genomic predictions than traditional EBV in purebred pigs. Genet. Sel. Evol. 2011, 43, 38. [Google Scholar] [CrossRef] [Green Version]
- Ge, F.; Jia, C.; Bao, P.; Wu, X.; Liang, C.; Yan, P. Accuracies of Genomic Prediction for Growth Traits at Weaning and Yearling Ages in Yak. Animals 2020, 10, 1793. [Google Scholar] [CrossRef] [PubMed]
- Pryce, J.E.; Arias, J.; Bowman, P.J.; Davis, S.R.; Macdonald, K.A.; Waghorn, G.C.; Wales, W.J.; Williams, Y.J.; Spelman, R.J.; Hayes, B.J. Accuracy of genomic predictions of residual feed intake and 250-day body weight in growing heifers using 625,000 single nucleotide polymorphism markers. J. Dairy Sci. 2011, 95, 2108–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean ± SE * | Minimum | Maximum |
|---|---|---|---|
| Birth weight (BW), kg | 34.48 ± 0.19 | 18.03 | 50.94 |
| Weaning weight (WW), kg | 377.98 ± 1.81 | 201.37 | 549.56 |
| Yearling weight (YW), kg | 540.67 ± 3.45 | 225.51 | 769.67 |
| Depth of rib fat (FAT), cm | 0.57 ± 0.01 | 0.02 | 1.40 |
| Intramuscular fat (IMF), % | 4.81 ± 0.04 | 2.02 | 9.77 |
| Longissimus muscle area (LMA), cm2 | 62.31 ± 0.41 | 27.22 | 91.19 |
| K-Means Cluster | |||||
| THE GBLUP | BayesA | BayesB | BayesC | BL | |
| Growth Traits | |||||
| BW | 0.23 [0.20, 0.26] | 0.09 [0.05, 0.13] | 0.10 [0.07, 0.13] | 0.09 [0.06, 0.11] | 0.15 [0.14, 0.16] |
| WW | 0.22 [0.19, 0.24] | 0.06 [0.02, 0.11] | 0.07 [0.02, 0.11] | 0.06 [0.05, 0.08] | 0.13 [0.12, 0.13] |
| YW | 0.32 [0.30, 0.36] | 0.16 [0.11, 0.22] | 0.16 [0.09, 0.20] | 0.15 [0.11, 0.19] | 0.17 [0.15, 0.18] |
| Carcass Traits | |||||
| FAT | 0.31 [0.28, 0.34] | 0.13 [0.09, 0.19] | 0.14 [0.09, 0.17] | 0.13 [0.10, 0.17] | 0.15 [0.14, 0.16] |
| IMF | 0.34 [0.31, 0.37] | 0.20 [0.16, 0.24] | 0.21 [0.15, 0.25] | 0.20 [0.16, 0.23] | 0.19 [0.18, 0.20] |
| LMA | 0.35 [0.30, 0.38] | 0.18 [0.13, 0.23] | 0.18 [0.11, 0.21] | 0.18 [0.12, 0.21] | 0.17 [0.15, 0.18] |
| Random Cluster | |||||
| GBLUP | BayesA | BayesB | BayesC | BL | |
| Growth Traits | |||||
| BW | 0.24 [0.20, 0.30] | 0.09 [0.04, 0.15] | 0.10 [0.04, 0.20] | 0.09 [0.06, 0.13] | 0.15 [0.14, 0.16] |
| WW | 0.21 [0.17, 0.24] | 0.06 [0.02, 0.17] | 0.06 [0.02, 0.11] | 0.06 [0.04, 0.09] | 0.12 [0.11, 0.13] |
| YW | 0.31 [0.26, 0.38] | 0.16 [0.08, 0.29] | 0.15 [0.08, 0.25] | 0.15 [0.11, 0.21] | 0.17 [0.15, 0.18] |
| Carcass Traits | |||||
| FAT | 0.30 [0.23, 0.35] | 0.14 [0.07, 0.23] | 0.14 [0.06, 0.23] | 0.13 [0.09, 0.18] | 0.15 [0.14, 0.16] |
| IMF | 0.34 [0.29, 0.39] | 0.21 [0.13, 0.28] | 0.21 [0.13, 0.31] | 0.20 [0.15, 0.25] | 0.19 [0.17, 0.20] |
| LMA | 0.35 [0.29, 0.42] | 0.19 [0.11, 0.29] | 0.18 [0.11, 0.28] | 0.18 [0.13, 0.21] | 0.17 [0.15, 0.18] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peters, S.O.; Kızılkaya, K.; Sinecen, M.; Mestav, B.; Thiruvenkadan, A.K.; Thomas, M.G. Genomic Prediction Accuracies for Growth and Carcass Traits in a Brangus Heifer Population. Animals 2023, 13, 1272. https://doi.org/10.3390/ani13071272
Peters SO, Kızılkaya K, Sinecen M, Mestav B, Thiruvenkadan AK, Thomas MG. Genomic Prediction Accuracies for Growth and Carcass Traits in a Brangus Heifer Population. Animals. 2023; 13(7):1272. https://doi.org/10.3390/ani13071272
Chicago/Turabian StylePeters, Sunday O., Kadir Kızılkaya, Mahmut Sinecen, Burcu Mestav, Aranganoor K. Thiruvenkadan, and Milton G. Thomas. 2023. "Genomic Prediction Accuracies for Growth and Carcass Traits in a Brangus Heifer Population" Animals 13, no. 7: 1272. https://doi.org/10.3390/ani13071272
APA StylePeters, S. O., Kızılkaya, K., Sinecen, M., Mestav, B., Thiruvenkadan, A. K., & Thomas, M. G. (2023). Genomic Prediction Accuracies for Growth and Carcass Traits in a Brangus Heifer Population. Animals, 13(7), 1272. https://doi.org/10.3390/ani13071272