A Fast and Powerful Empirical Bayes Method for Genome-Wide Association Studies

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Statistical Model for Marker Scanning
2.2. The Modified Kinship Matrix
2.3. Parameter Estimation and a Special Algorithm for Fast Computation
2.4. Hypothesis Test
2.5. EMMA and EB
2.6. Simulation Studies
2.6.1. Experiment 1
2.6.2. Experiment 2
2.7. Beef Cattle Data
2.7.1. Ethics Statement
2.7.2. Animals and Phenotypes
2.7.3. Genotype Data and Quality Control
3. Results
3.1. Simulation Study
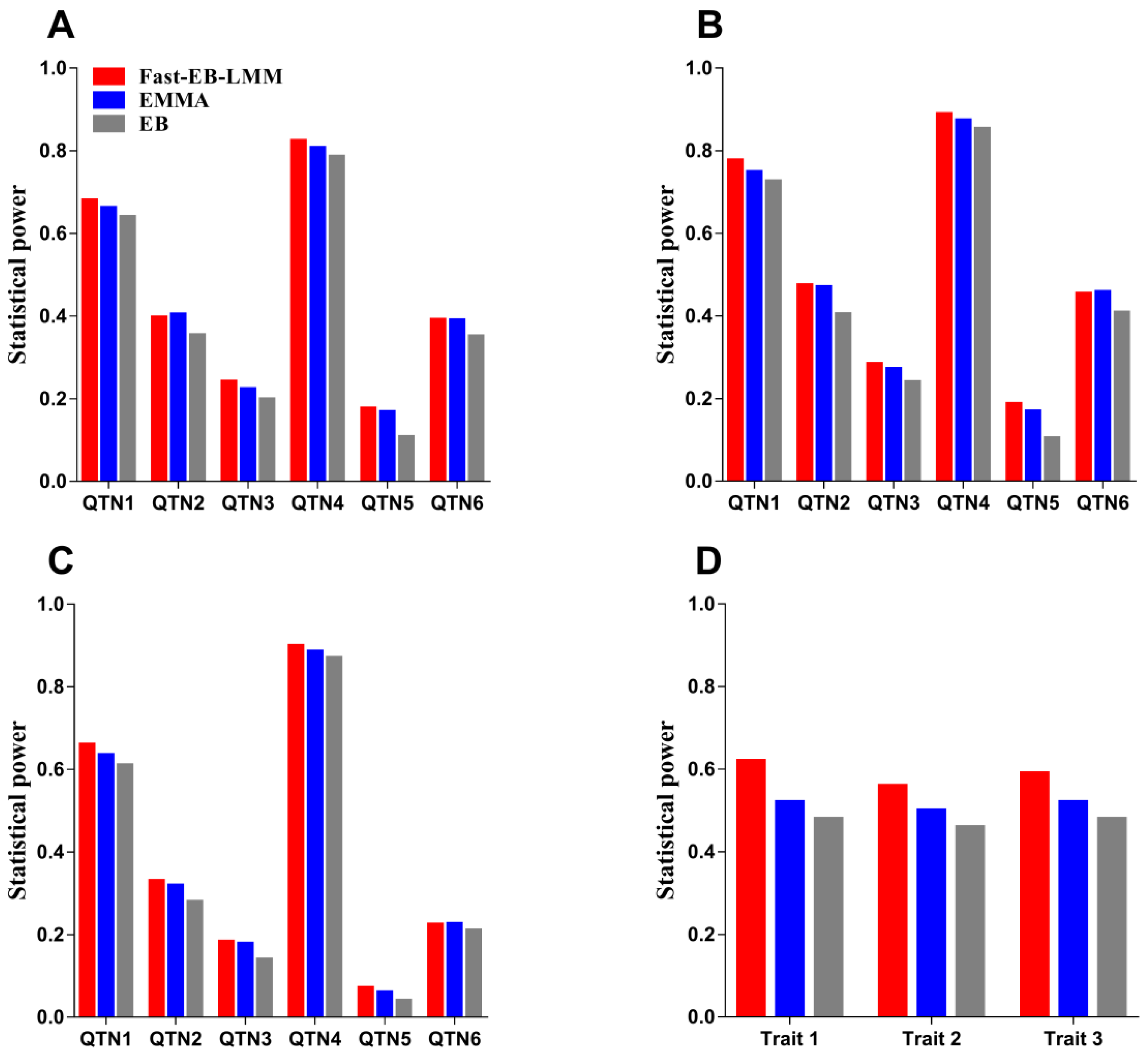
3.1.1. Statistical Power for QTN Detection
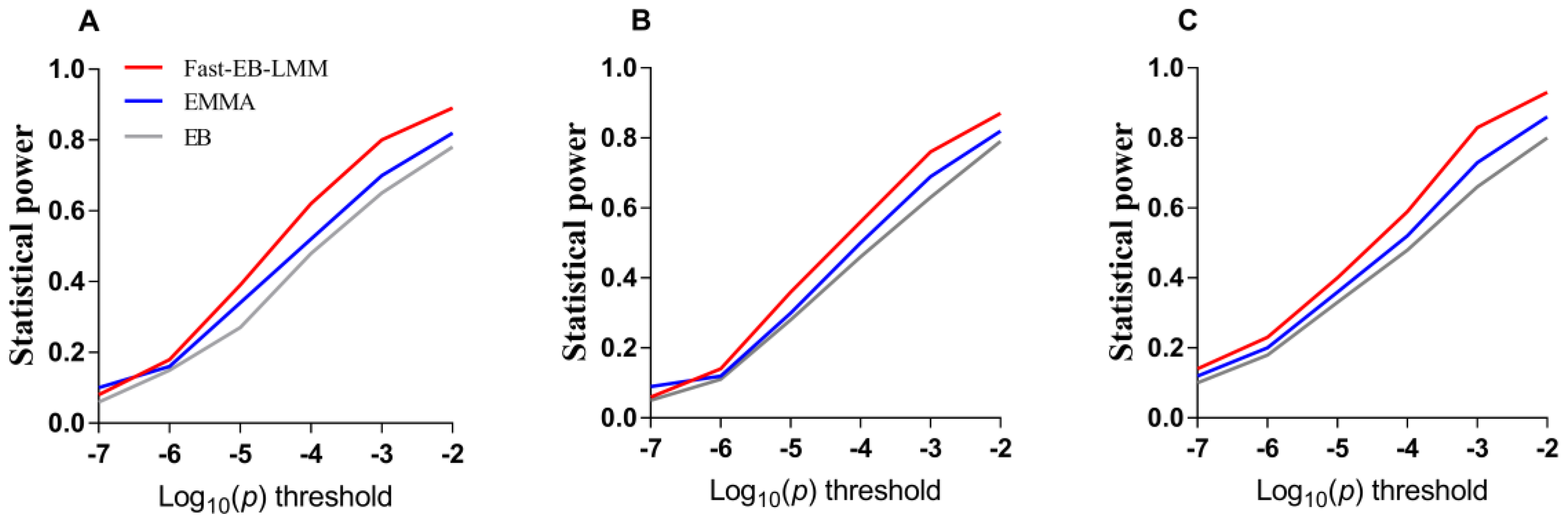
3.1.2. False Positive Rate and ROC Curve
3.1.3. Computational Efficiency
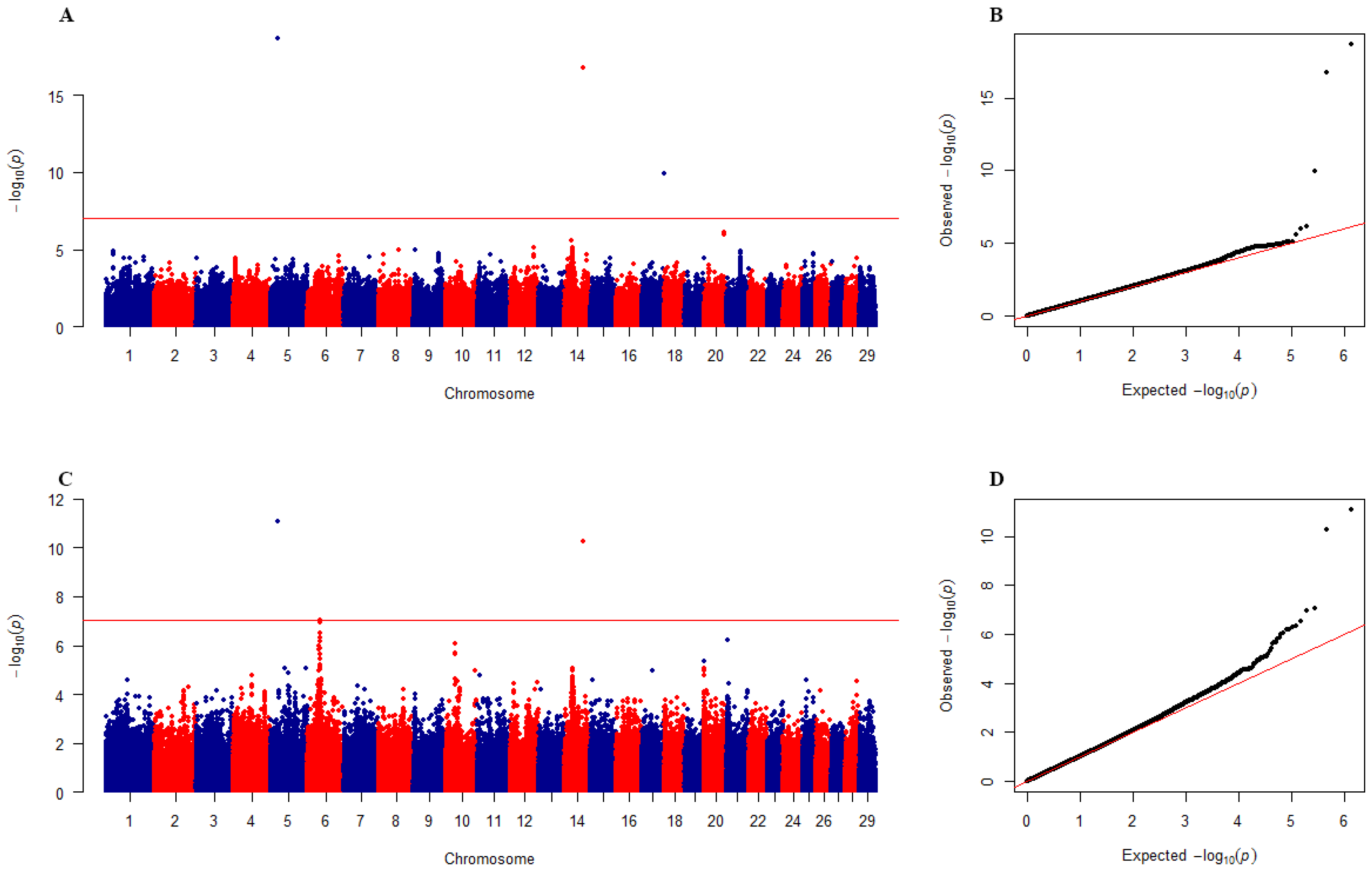
3.2. Beef Cattle Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.W.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.M.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Na. Genet. 2010, 42, 355. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zaitlen, N.A.; Goddard, M.E.; Visscher, P.M.; Price, A.L. Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet. 2014, 46, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.M.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient control of population structure in model organism association mapping. Genetics 2008, 178, 1709–1723. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 2014, 11, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Lippert, C.; Listgarten, J.; Liu, Y.; Kadie, C.M.; Davidson, R.I.; Heckerman, D. FaST linear mixed models for genome-wide association studies. Nat. Methods 2011, 8, 833–835. [Google Scholar] [CrossRef]
- Wang, Q.; Wei, J.; Pan, Y.; Xu, S. An efficient empirical Bayes method for genomewide association studies. J. Anim. Breed. Genet. 2016, 133, 253–263. [Google Scholar] [CrossRef]
- Aulchenko, Y.S.; de Koning, D.J.; Haley, C. Genomewide rapid association using mixed model and regression: A fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics 2007, 177, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Vilhjalmsson, B.J.; Nordborg, M. The nature of confounding in genome-wide association studies. Nat. Rev. Genet. 2013, 14, 1–2. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Yang, J.; Chen, G.B.; Ripke, S.; Stahl, E.A.; Hultman, C.M.; Sklar, P.; Visscher, P.M.; Sullivan, P.F.; Goddard, M.E.; et al. Estimation of SNP heritability from dense genotype data. Am. J. Hum. Genet. 2013, 93, 1151–1155. [Google Scholar] [CrossRef]
- Listgarten, J.; Lippert, C.; Kadie, C.M.; Davidson, R.I.; Eskin, E.; Heckerman, D. Improved linear mixed models for genome-wide association studies. Nat. Methods 2012, 9, 525–526. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.R. Best linear unbiased estimation and prediction under a selection model. Biometrics 1975, 31, 423–447. [Google Scholar] [CrossRef] [PubMed]
- Xu, S. An empirical Bayes method for estimating epistatic effects of quantitative trait loci. Biometrics 2007, 63, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.J.; Zhang, H.; Ni, Y.L.; Huang, B.; Zhang, J.; Feng, J.Y.; Wang, S.B.; Dunwell, J.M.; Zhang, Y.M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef]
- Atwell, S.; Huang, Y.S.; Vilhjalmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.M.; Hu, T.T.; Jiang, R.; et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Usai, M.G.; Gaspa, G.; Macciotta, N.P.; Carta, A.; Casu, S. XVI(th) QTLMAS: Simulated dataset and comparative analysis of submitted results for QTL mapping and genomic evaluation. BMC Proc. 2014, 8, S1. [Google Scholar] [CrossRef] [PubMed]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Chang, T.; Xia, J.; Xu, L.; Wang, X.; Zhu, B.; Zhang, L.; Gao, X.; Chen, Y.; Li, J.; Gao, H. A genome-wide association study suggests several novel candidate genes for carcass traits in Chinese Simmental beef cattle. Anim. Genet. 2018, 49, 312–316. [Google Scholar] [CrossRef]
- Miao, J.; Wang, X.; Bao, J.; Jin, S.; Chang, T.; Xia, J.; Yang, L.; Zhu, B.; Xu, L.; Zhang, L.; et al. Multimarker and rare variants genomewide association studies for bone weight in Simmental cattle. J. Anim. Breed. Genet. 2018, 135, 159–169. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Trait | Number | Mean a | SD a | h2 b | Phenotypic Correlation | |
|---|---|---|---|---|---|---|
| CW | BW | |||||
| CW | 1217 | 271.3 | 45.63 | 0.38 | 1 | - |
| BW | 1217 | 40.7 | 6.52 | 0.41 | 0.67 | 1 |
| Method | Simulation 1 | Simulation 2 | ||
|---|---|---|---|---|
| a | b | c | ||
| Fast-EB-LMM | 4.17 | 4.23 | 4.19 | 0.1 |
| EMMA | 13.82 | 14.04 | 13.91 | 0.31 |
| EB | 6.21 | 6.31 | 6.24 | 0.15 |
| Trait | SNP Name | BTA | Position(bp) a | p-Value b | Nearest Gene c | Distance d |
|---|---|---|---|---|---|---|
| CW | BovineHD0500006528 | 5 | 22,558,100 | 2.06E-19 | C12ORF74 | 161123 |
| BovineHD1400017455 | 14 | 62769117 | 1.76E-17 | RIMS2 | within | |
| BovineHD1700021340 | 17 | 73007522 | 1.12E-10 | BT.88981 | 3479 | |
| BW | BovineHD0500006528 | 5 | 22558100 | 8.17E-11 | C12ORF74 | 161,123 |
| BovineHD0600010952 | 6 | 39990876 | 8.82E-08 | LCORL | 998873 | |
| BovineHD0600010956 | 6 | 39997880 | 1.13E-07 | LCORL | 1005877 | |
| BovineHD1400017455 | 14 | 62769117 | 5.47E-11 | RIMS2 | within |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, T.; Wei, J.; Liang, M.; An, B.; Wang, X.; Zhu, B.; Xu, L.; Zhang, L.; Gao, X.; Chen, Y.; et al. A Fast and Powerful Empirical Bayes Method for Genome-Wide Association Studies. Animals 2019, 9, 305. https://doi.org/10.3390/ani9060305
Chang T, Wei J, Liang M, An B, Wang X, Zhu B, Xu L, Zhang L, Gao X, Chen Y, et al. A Fast and Powerful Empirical Bayes Method for Genome-Wide Association Studies. Animals. 2019; 9(6):305. https://doi.org/10.3390/ani9060305
Chicago/Turabian StyleChang, Tianpeng, Julong Wei, Mang Liang, Bingxing An, Xiaoqiao Wang, Bo Zhu, Lingyang Xu, Lupei Zhang, Xue Gao, Yan Chen, and et al. 2019. "A Fast and Powerful Empirical Bayes Method for Genome-Wide Association Studies" Animals 9, no. 6: 305. https://doi.org/10.3390/ani9060305
APA StyleChang, T., Wei, J., Liang, M., An, B., Wang, X., Zhu, B., Xu, L., Zhang, L., Gao, X., Chen, Y., Li, J., & Gao, H. (2019). A Fast and Powerful Empirical Bayes Method for Genome-Wide Association Studies. Animals, 9(6), 305. https://doi.org/10.3390/ani9060305