



Animals 2024, 14(7), 1098; https://doi.org/10.3390/ani14071098 - 4 Apr 2024
Viewed by 1299
Abstract
►
Show Figures
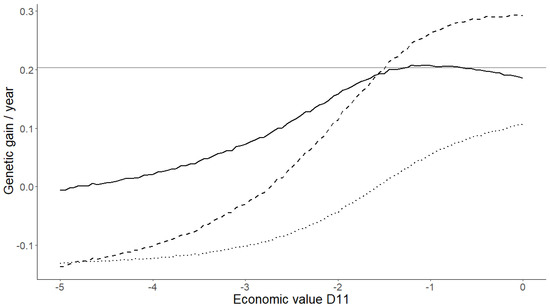
The need for sufficient reference population data poses a significant challenge in breeding programs aimed at improving pig farming on a small to medium scale. To overcome this hurdle, investigating the advantages of combing reference populations of varying sizes is crucial for enhancing
[...] Read more.
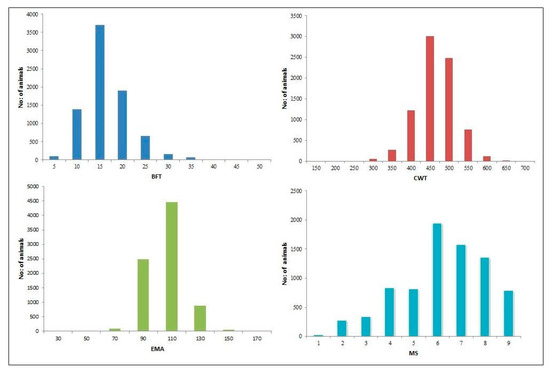
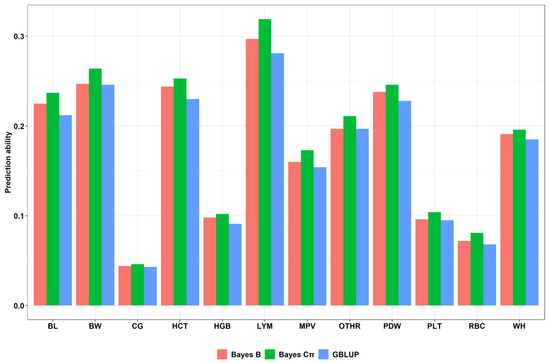
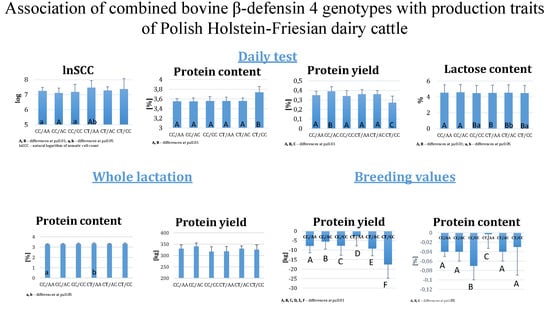
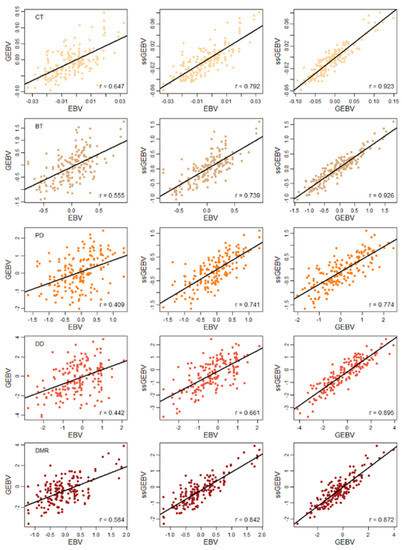
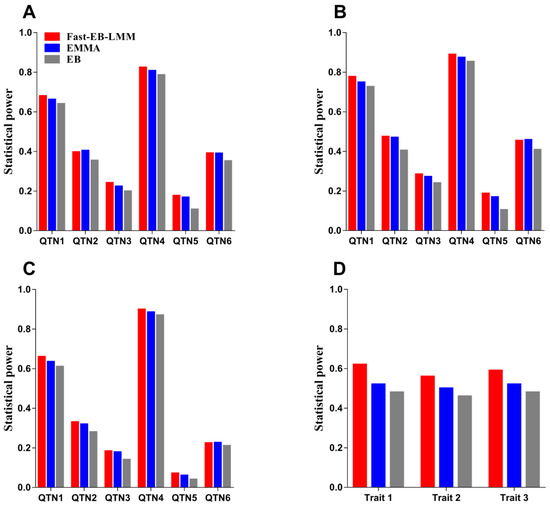
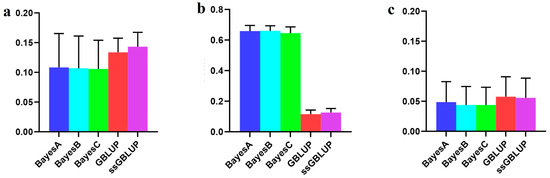
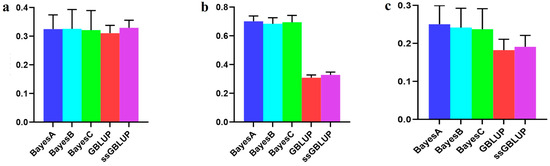
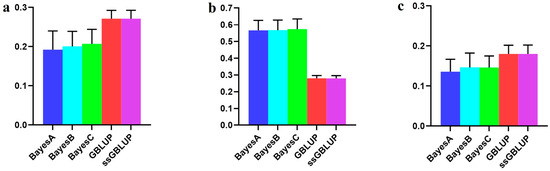
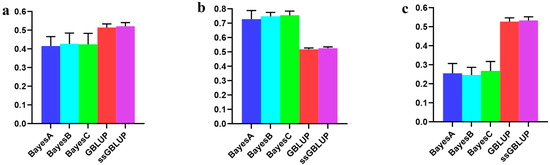
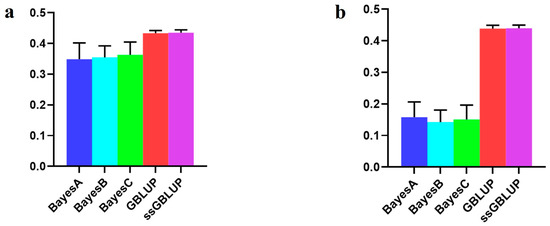
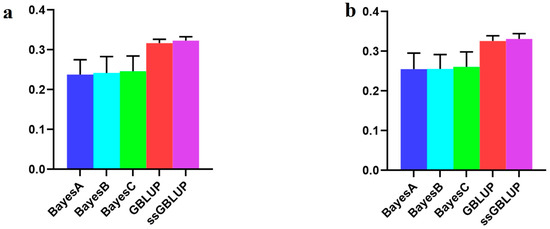
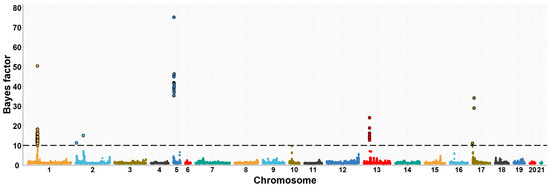
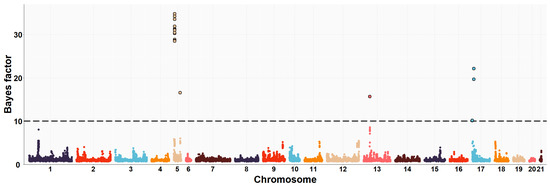
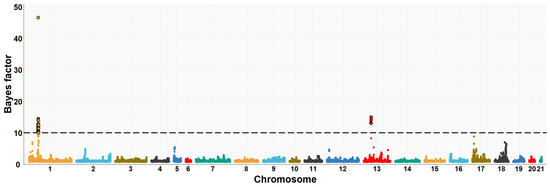
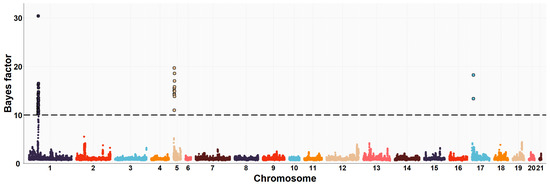
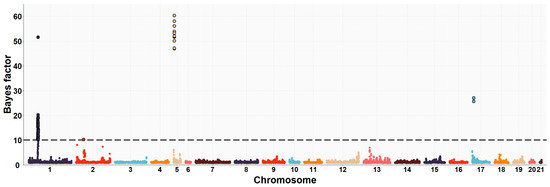
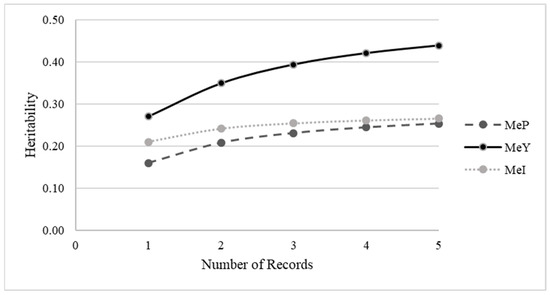
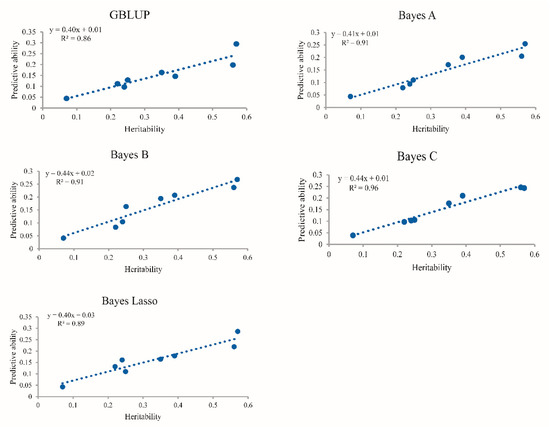
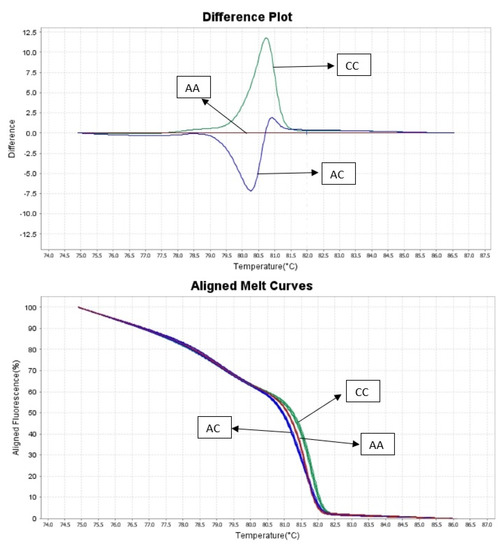
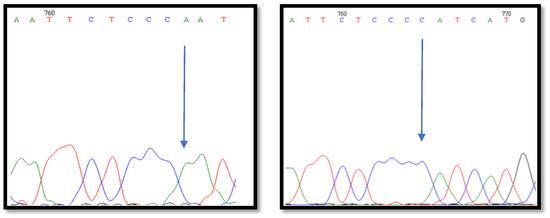
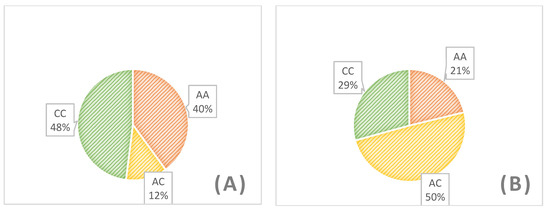
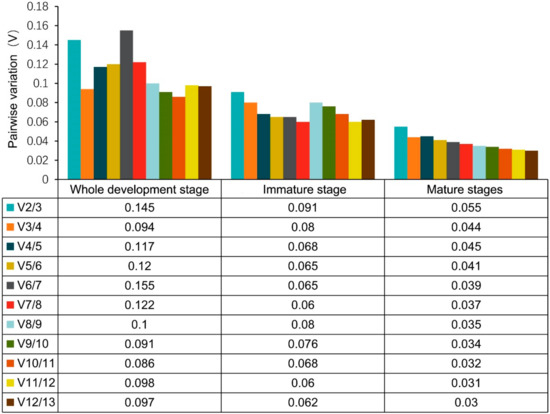
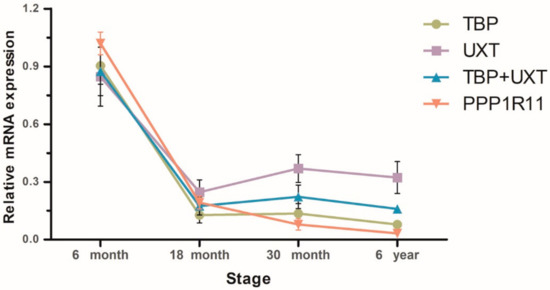
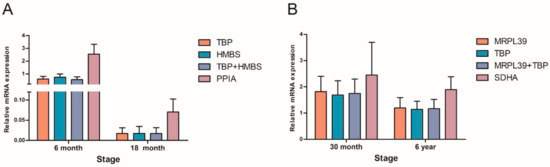
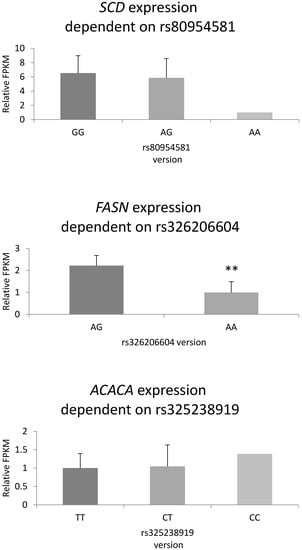
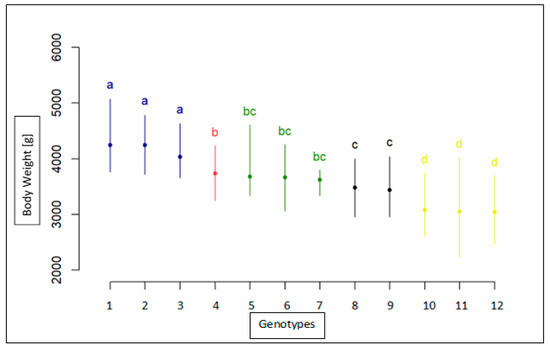
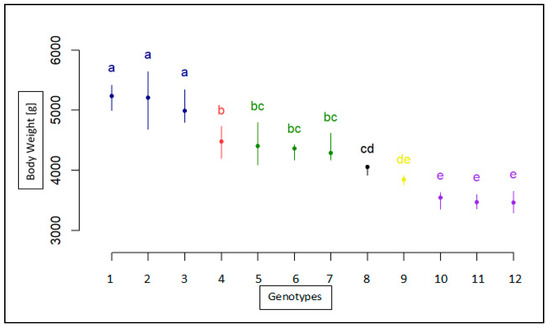
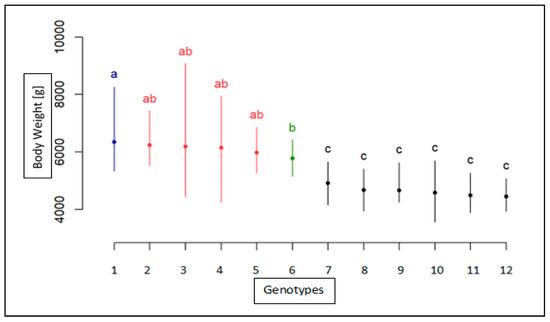
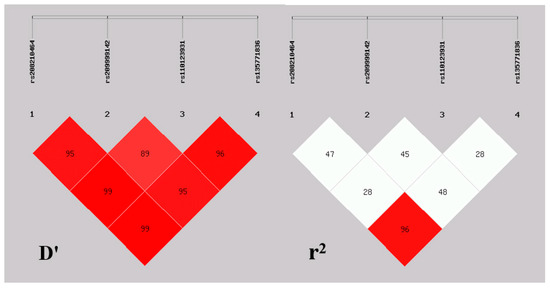
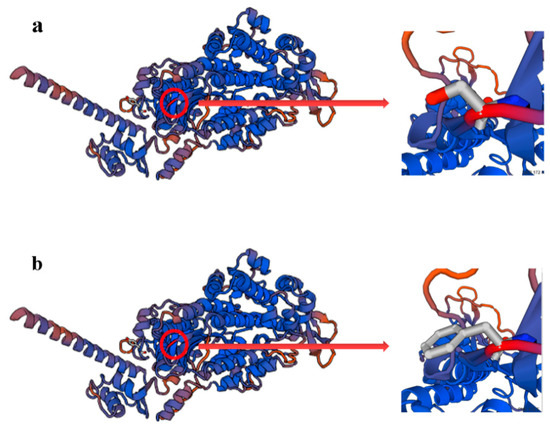
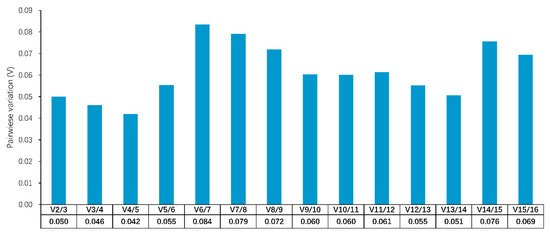
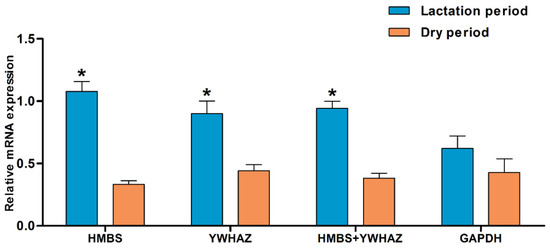
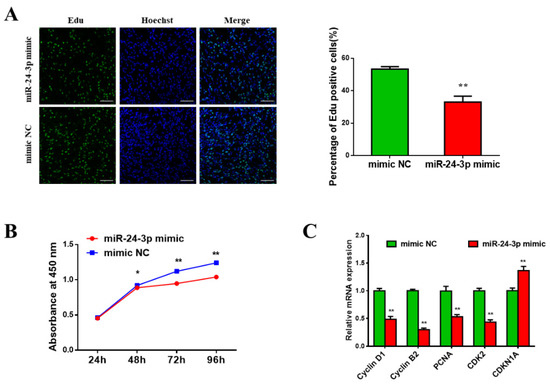
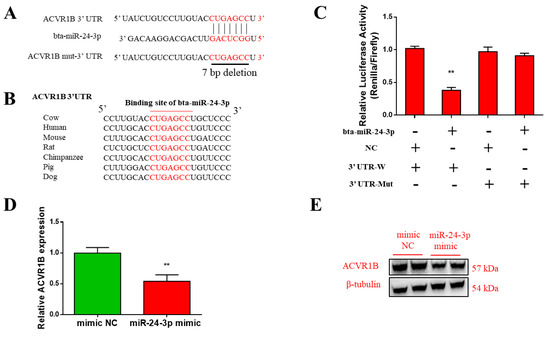
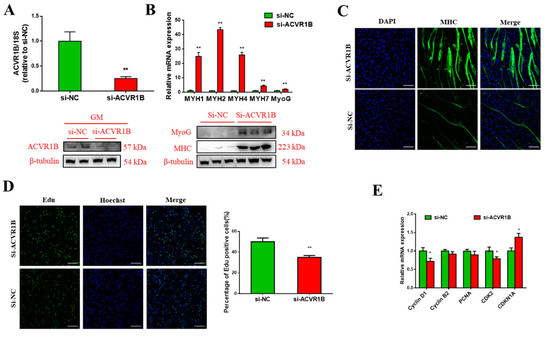
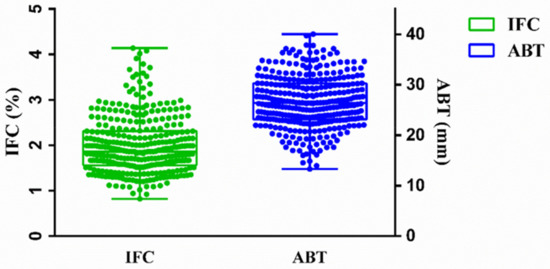
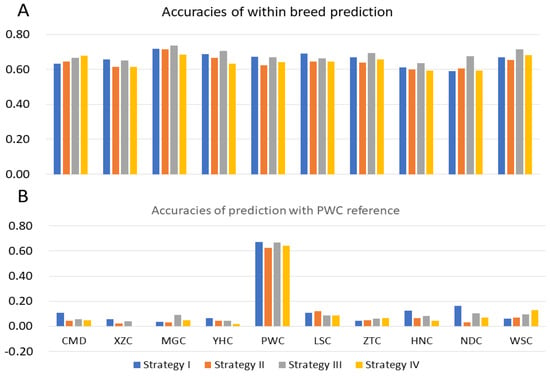
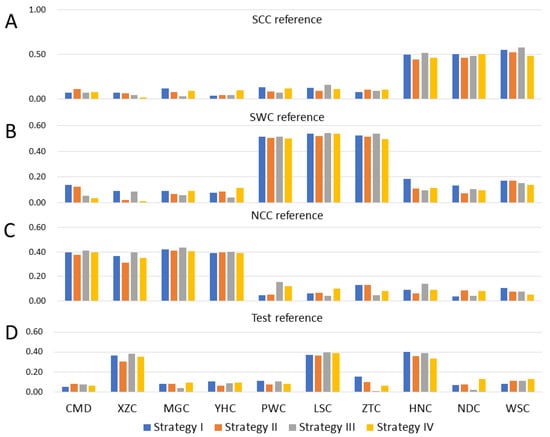
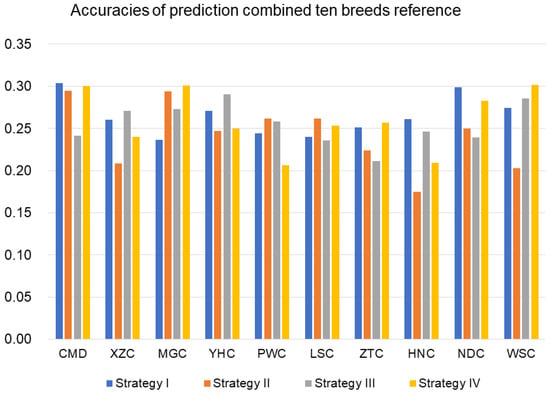
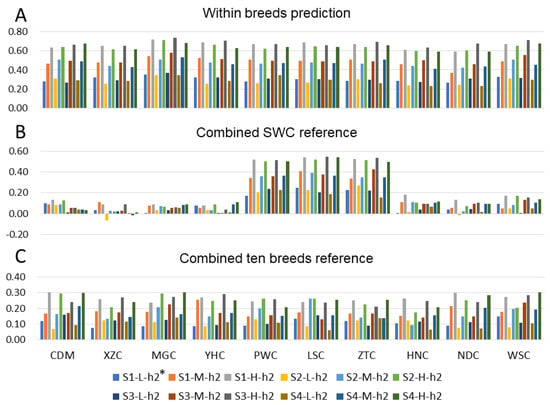
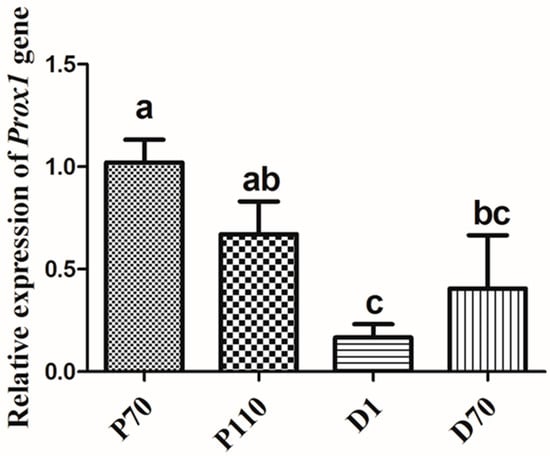
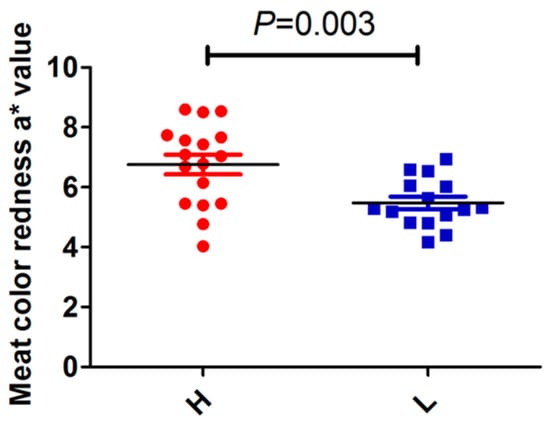
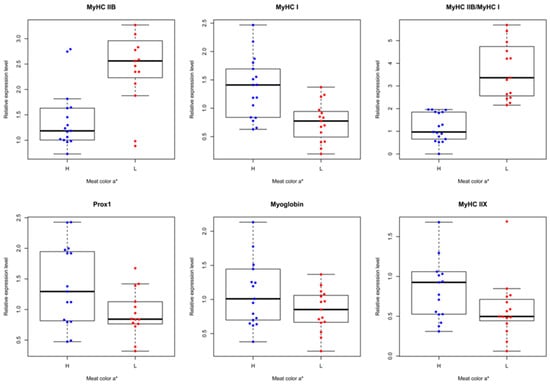
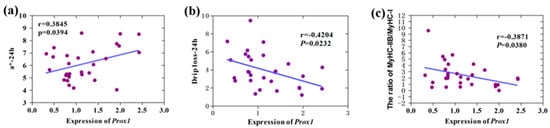
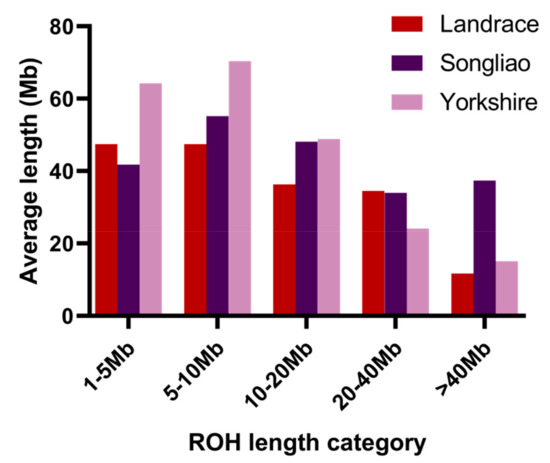
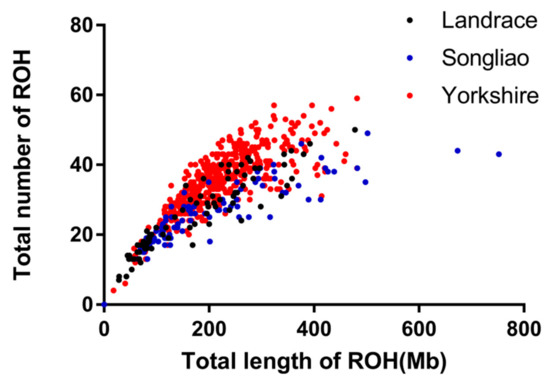
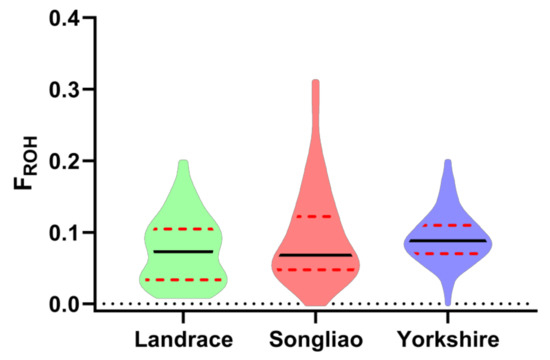
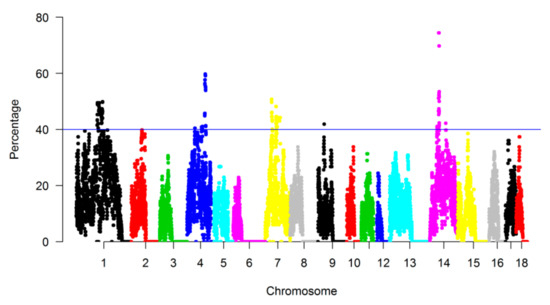
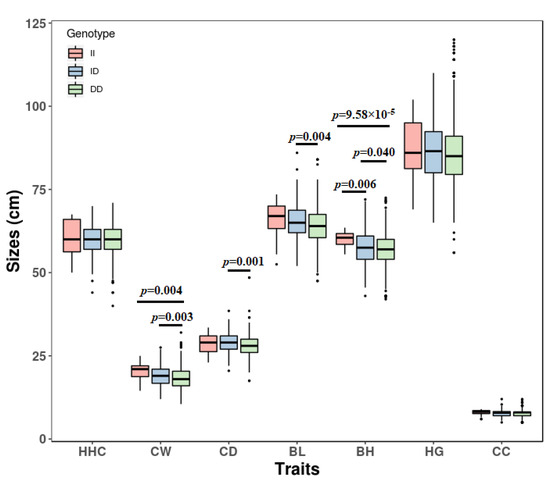
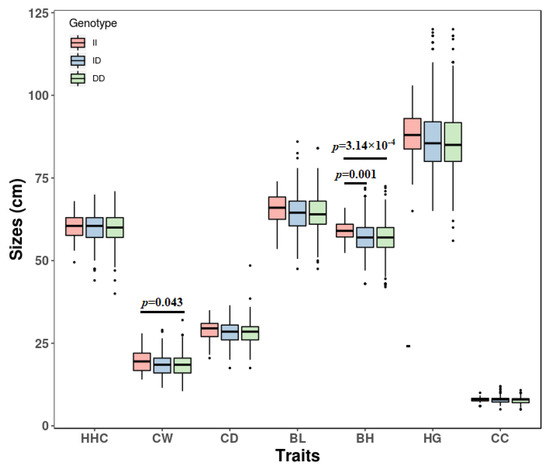
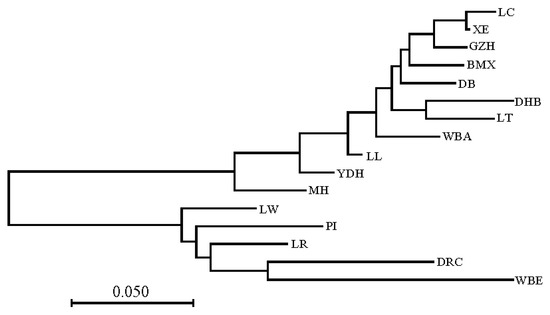
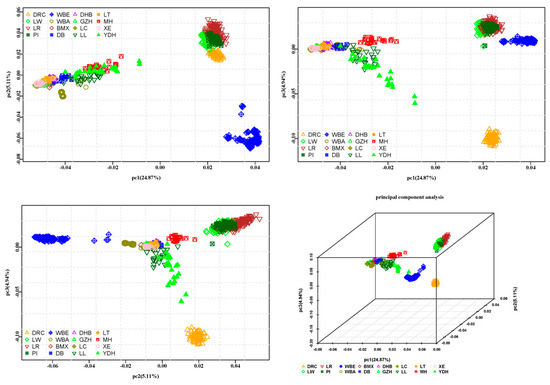
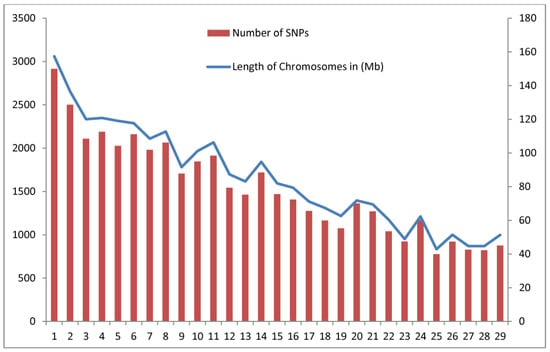
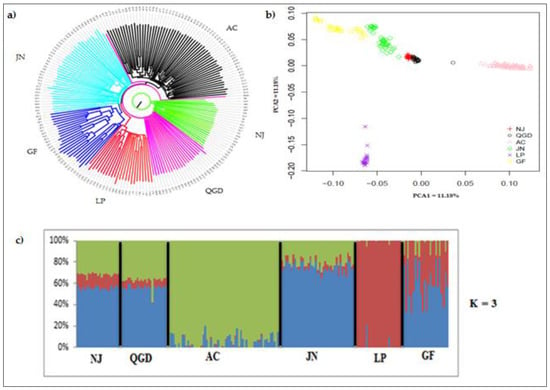
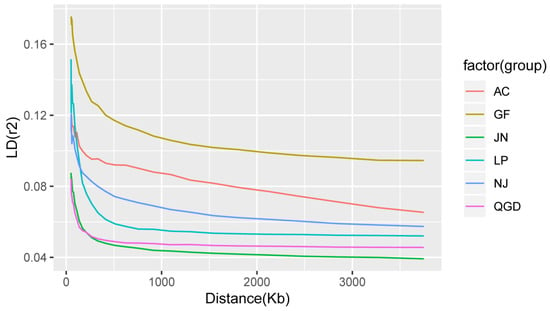
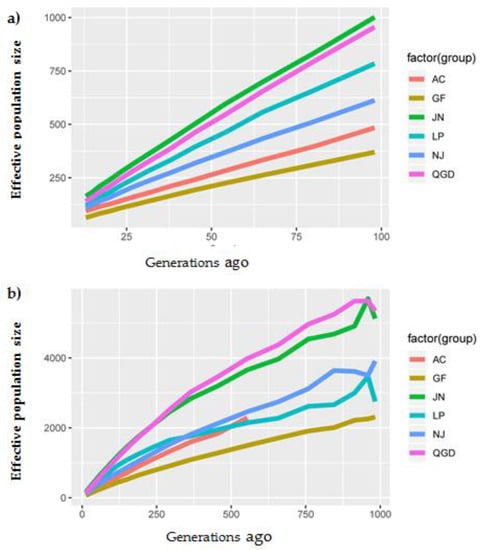
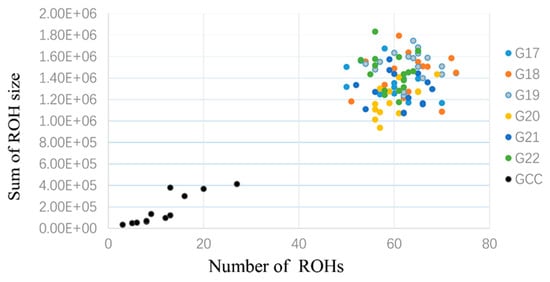
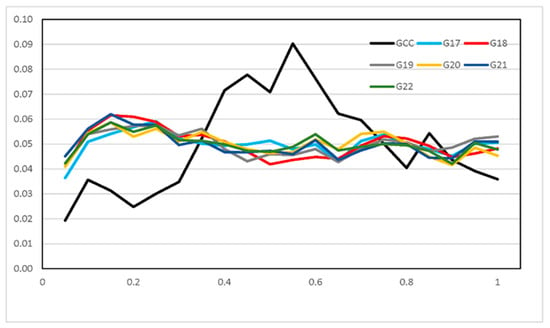
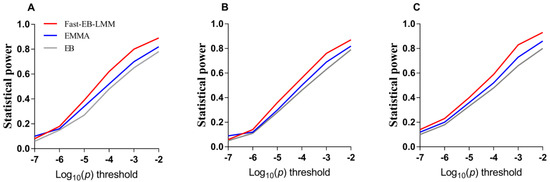
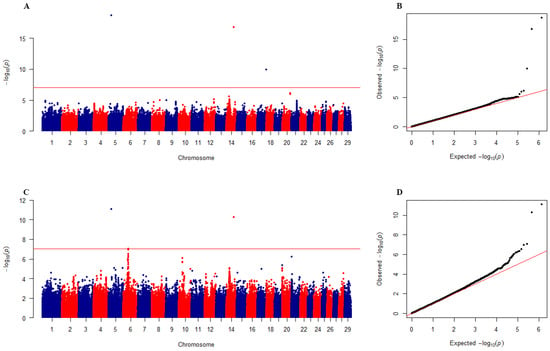
The need for sufficient reference population data poses a significant challenge in breeding programs aimed at improving pig farming on a small to medium scale. To overcome this hurdle, investigating the advantages of combing reference populations of varying sizes is crucial for enhancing the accuracy of the genomic estimated breeding value (GEBV). Genomic selection (GS) in populations with limited reference data can be optimized by combining populations of the same breed or related breeds. This study focused on understanding the effect of combing different reference group sizes on the accuracy of GS for determining the growth effectiveness and percentage of lean meat in Yorkshire pigs. Specifically, our study investigated two important traits: the age at 100 kg live weight (AGE100) and the backfat thickness at 100 kg live weight (BF100). This research assessed the efficiency of genomic prediction (GP) using different GEBV models across three Yorkshire populations with varying genetic backgrounds. The GeneSeek 50K GGP porcine high-density array was used for genotyping. A total of 2295 Yorkshire pigs were included, representing three Yorkshire pig populations with different genetic backgrounds—295 from Danish (small) lines from Huaibei City, Anhui Province, 500 from Canadian (medium) lines from Lixin County, Anhui Province, and 1500 from American (large) lines from Shanghai. To evaluate the impact of different population combination scenarios on the GS accuracy, three approaches were explored: (1) combining all three populations for prediction, (2) combining two populations to predict the third, and (3) predicting each population independently. Five GEBV models, including three Bayesian models (BayesA, BayesB, and BayesC), the genomic best linear unbiased prediction (GBLUP) model, and single-step GBLUP (ssGBLUP) were implemented through 20 repetitions of five-fold cross-validation (CV). The results indicate that predicting one target population using the other two populations yielded the highest accuracy, providing a novel approach for improving the genomic selection accuracy in Yorkshire pigs. In this study, it was found that using different populations of the same breed to predict small- and medium-sized herds might be effective in improving the GEBV. This investigation highlights the significance of incorporating population combinations in genetic models for predicting the breeding value, particularly for pig farmers confronted with resource limitations.
Full article
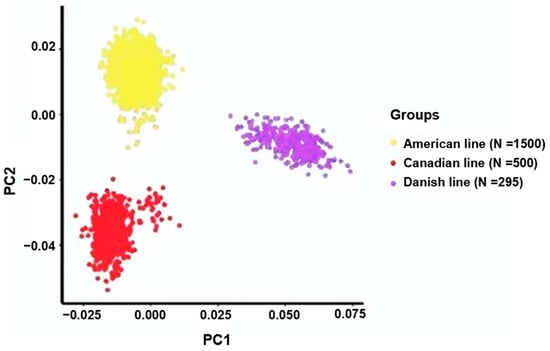
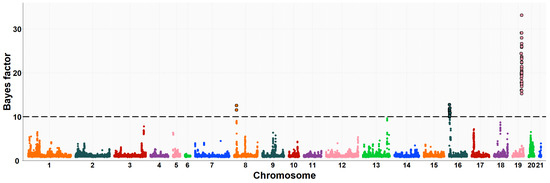
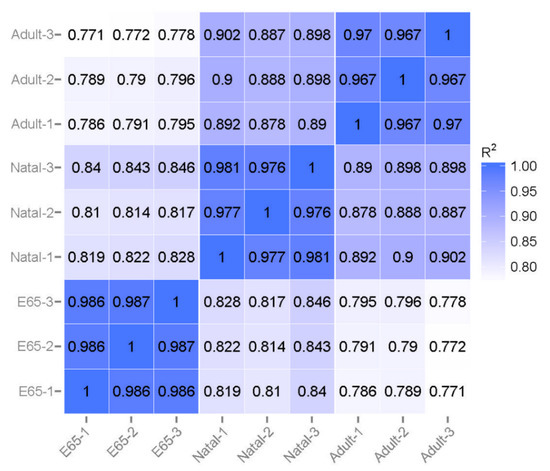
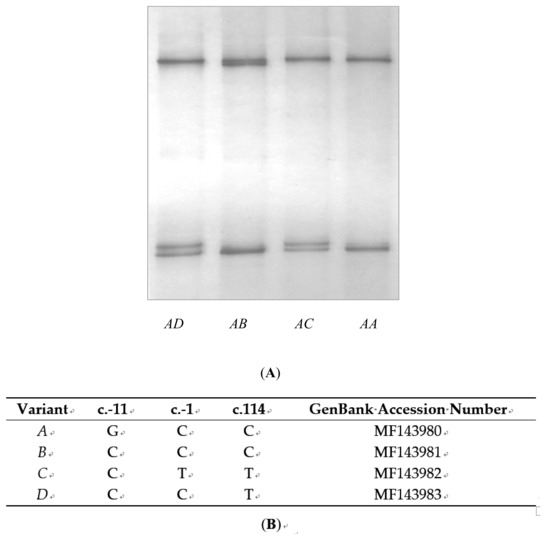
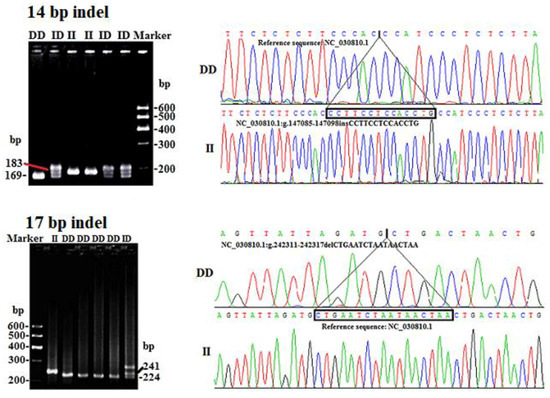
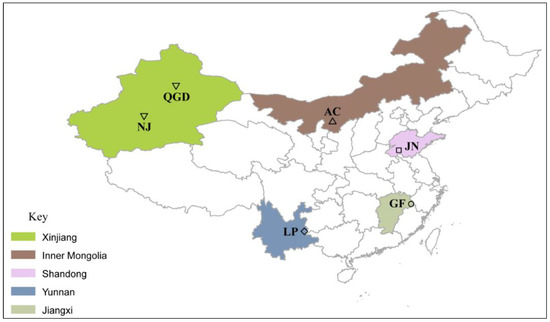
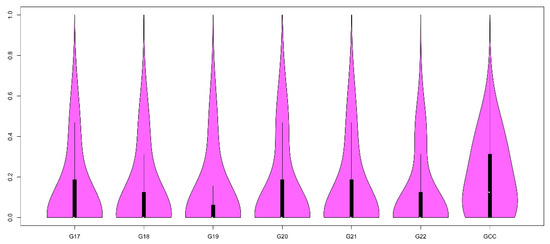
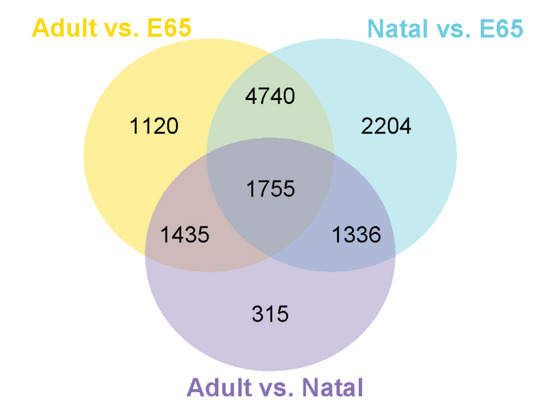
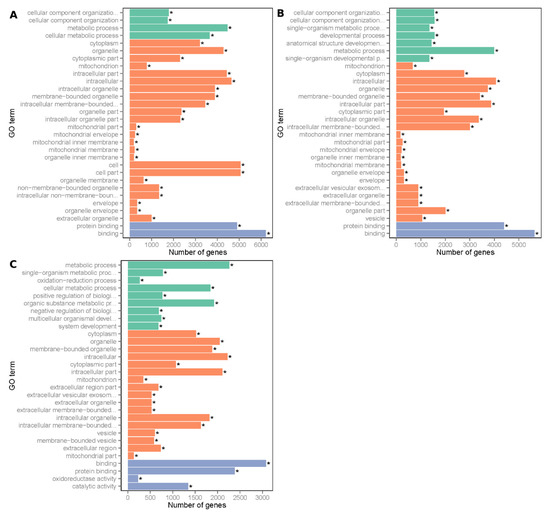
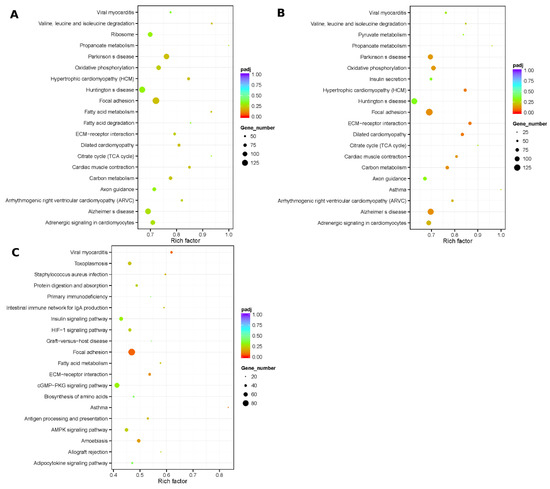
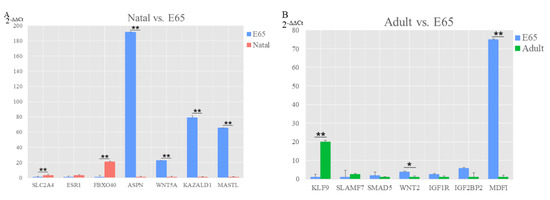
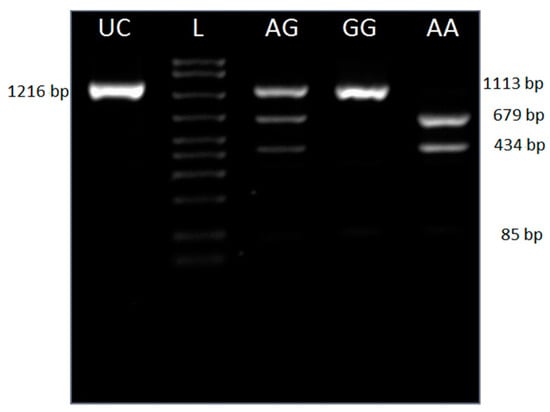
Figure 1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}