4.1. Evaluation of the Optimal Crystallization Heat Treatment
Evaluation of the suitable crystallization regime is essential for the successful conversion of the initial glass into a fine crystalline glass-ceramic. Typically, the times and temperatures for nucleation and crystallization stages are estimated by sophisticated microscopic studies and quantitative XRD analysis [
1,
24], which, unfortunately, are expensive and time-consuming. However, other techniques can also be applied.
The most popular alternative for estimating nucleation temperature and time is use of non-isothermal DTA experiments [
12,
24]. In this case, the temperature shifts, ΔT, of the exothermic crystallization peak temperature, T
p, to inferior temperatures is used as an indicator for the effectiveness of the nucleation process. This is a consequence of the fact that the formation of a significant number of new nuclei can result in a smaller size of crystals being formed, which accelerates the rate of crystallization. The higher the number of formed crystals, the smaller their size and the larger the temperature shift.
The DTA method is also widely used to estimate the crystallization tendency (i.e., bulk or surface). In this case, the ability for bulk nucleation and growth is evaluated by a simple comparison of the DTA runs of bulk and fine powder samples. At a satisfactory tendency for bulk crystallization, the Tp of the bulk sample can be similar to that of the powder sample. To the contrary, when the tendency for bulk nucleation is scarce, the crystallization exo-peak is larger, with lower intensity, and it appears at a higher temperature.
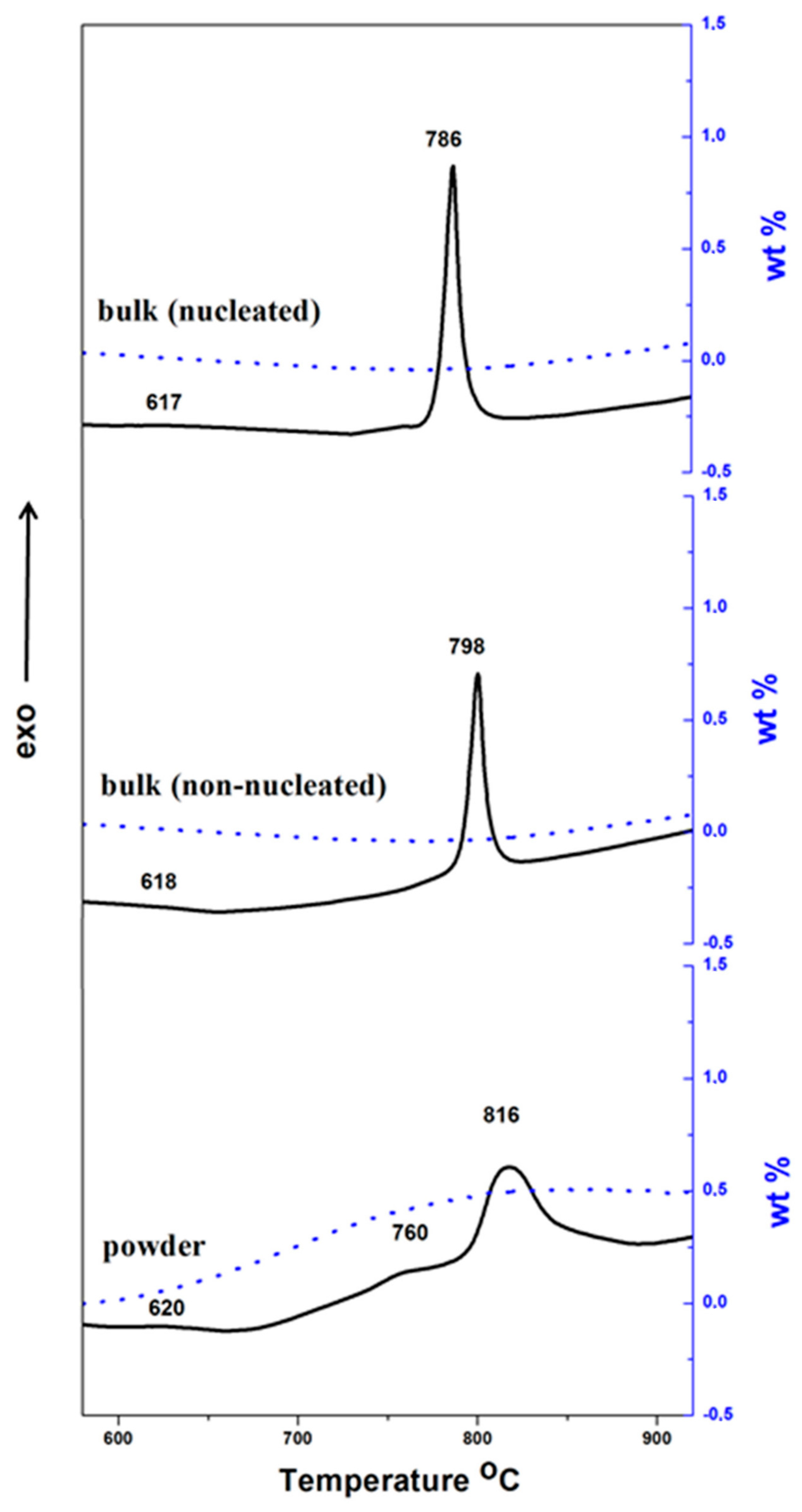
The DTA–TG data of the powder sample (below 75 µm) and that for non-nucleated and nucleated bulk samples of the discussed glass are shown in
Figure 2. The DTA results elucidate a glass transition temperature of 615–620 °C (estimated as the first onset of the heat flow signal in the glass transition range) for all samples, and T
p at ~816, ~798 and 786 °C for powder sample, non-nucleated and nucleated bulk samples, respectively. Additionally, the traces of the powder sample show an exotherm with low intensity in the interval 720–780 °C, which is connected with a weight increase of about 0.5 wt %. This effect can be associated with the surface oxidation of Fe
2+ into Fe
3+ which takes place before the crystallization in the glass powder. Similar DTA peaks and TG signals are not present in the bulk samples due to their low specific surfaces. Since a complete oxidation of the iron oxides is expected in the glass transition temperature interval, the value of TG gain might be used to evaluate the ratio Fe
2+/Fe
3+ in the parent glass [
9,
14]. For our composition, the weight rise due to FeO oxidation is ~0.55 wt %, which corresponds to an initial FeO amount of ~5% and a FeO/F
2O
3 ratio of ~0.34. This ratio is typical for similar glass compositions and melting procedures and is not very different from the magnetite (FeO·Fe
2O
3) ratio of 0.5, one which can provoke the most exhaustive formation of iron spinel.
The higher crystallization temperature of the powder samples is unusual behavior, consequent to the surface oxidation Fe
2+ into Fe
3+, leading to an increase of viscosity and to a significant decrease of the magnetite (FeO·Fe
2O
3) spinel [
5] formation in the glass powders. The viscosity variation is result of the different structural roles of Fe
2+ (which is a typical “modifier”) and Fe
3+ (acting as “intermediates”) in glass and melt structures.
The intense DTA peak of the non-nucleated bulk sample at 798 °C, only about 200 °C higher than the glass transition temperature, indicates a notable bulk crystallization, even without nucleation heat-treatment. As has been already noted, this behavior can be considered typical for glasses rich of iron oxides. Nevertheless, the nucleation treatment additionally decreases the crystallization temperature, which can lead to a finer crystalline structure.
The optimum nucleation temperature was defined with DTA experiments at 20 °C/min by means of bulk particles, heat-treated previously for 1 h at diverse temperatures in the expected “nucleation” range (T
g − 20 °C)–(T
g + 50 °C). The T
p of observed exotherms and the corresponding ΔT shifts are reported in
Table 3. It is evident that the optimum temperature for nucleation is at about 630–650 °C, where ΔT is 11–12 °C. In the glass transition range, practically no temperature shift was noted, while at 615 and at 670 °C ΔT was only 3–4 °C. Similarly, the optimum nucleation time at 650 °C was also obtained. These results also are presented in
Table 3 and show that the maximum temperature shift was reached after only 45–60 min.
The temperature and time of crystallization were estimated by pycnometric measurements. In these experiments, both nucleated specimens (heat-treated for 1 h at 650 ± 5 °C) and non-nucleated specimens, weighing about 2–3 g were heat-treated at 710 ± 5 °C, 730 ± 5 °C, 750 ± 5 °C and 770 ± 5 °C for times between 1 and 120 min. The heat treatments were carried out in an electric furnace at 10 °C/min heating and 20 °C/min cooling. The densities of each sample were measured before and after the crystallization. The density of the initial glass samples varied in the range of 2.86–2.87 g/cm3, whereas the maximum density reached was 3.17–3.18 g/cm3.
The changes in density (Δρ) of nucleated and non-nucleated samples treated at 730 °C for different times are reported in
Table 4a. In both series, during the first 20 min the phase formation is faster, and then the rate of crystallization begins to decrease. The results also clarify that the crystallization process is significantly accelerated in the nucleated samples, and that the increase in crystallinity becomes negligible after 1 h holding.
The density variations and the corresponding amounts of crystal phase formed after 5 min holding at diverse temperature are summarized in Figure 4b. The results support the proposition that the preliminary nucleation treatment accelerates the crystallization and that the temperature significantly influences the crystallization rate: at 770 °C, the maximum crystallinity of 52–54% is reached after only 5–10 min, while at 750 °C, the maximum crystallinity of 55–57% is obtained after 30–40 min; at 710 °C the formed-crystal phase remains below 10–13%, even after 2 h of holding. The observed increase of the significant amount of formed-crystal phase with the decrease of temperature (i.e., 59–61% at 730 °C vs. 52–54% at 770 °C) can be explained by the diminishing of precipitation with rises in temperature.
These results demonstrate that the optimum temperature for the crystallization step is 730–740 °C, while the crystallization hold is about 45–60 min. If the temperature is lower with 20–30 °C, the crystallization time will increase significantly, whereas at higher temperature (760–770 °C) a distinguished decrease of the crystallinity occurs. In addition, if the crystallization rate is too high, some problems with the formation of regular structure can be expected due to the intensive release of latent crystallization heat.
4.2. Phase Formation and Structure
The efficiency of the optimal thermal regime was studied using XRD, SEM, TEM and FESEM. Various samples, obtained with and without the nucleation step at 650 °C for 1 h, and with 1, 5 and 60 min crystallization holds at 730 °C, were made and analyzed. These specimens were labeled GC0-1, GC0-5, GC0-60, GC60-1, GC60-5 and GC60-60; the first number indicates the nucleation time, and the second, the crystallization time.
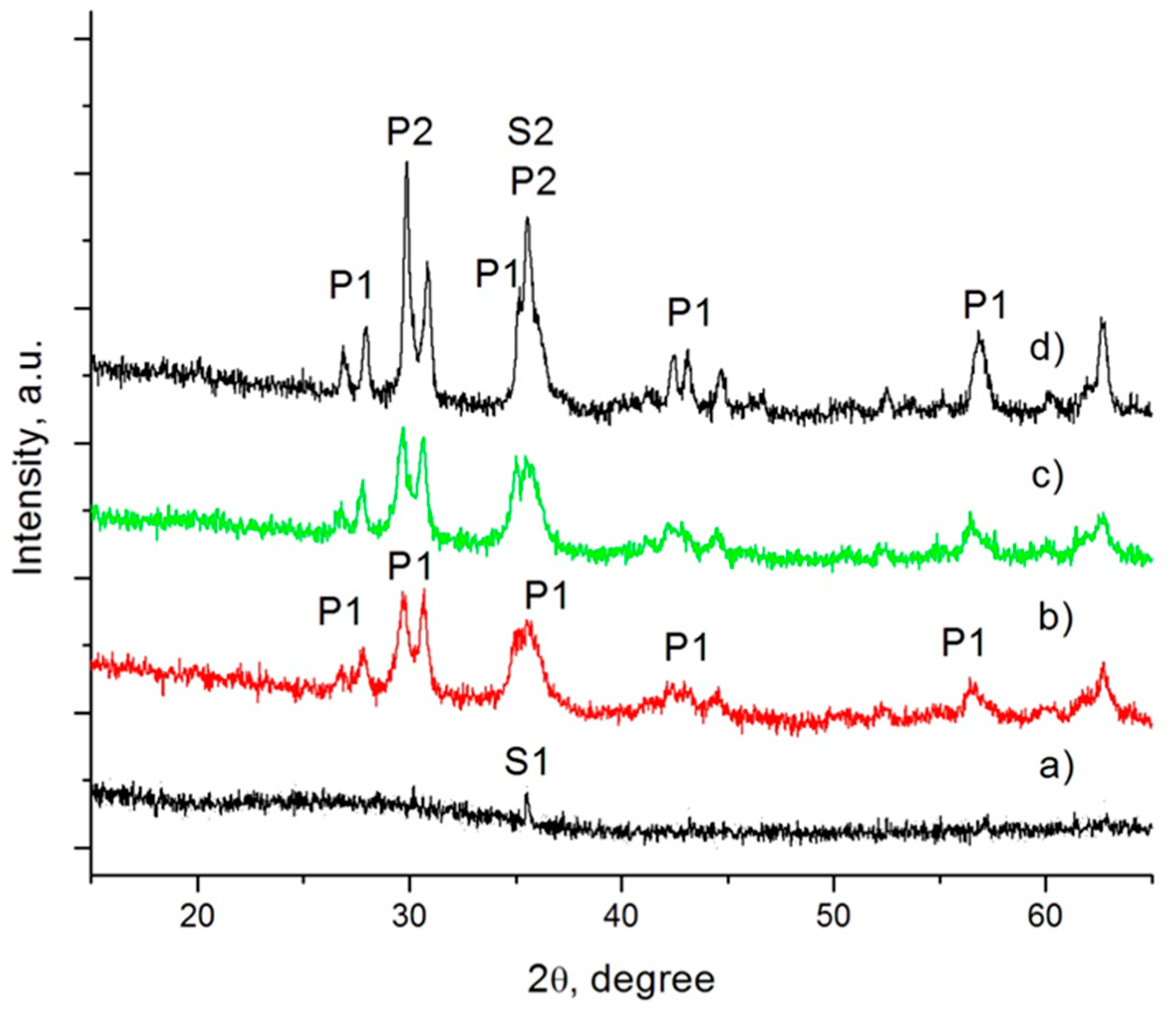
The XRD pattern of the parent glass (a) and specimens GC60-1 (b), GC60-5 (c) and GC60-60 (d) are shown in
Figure 3.
Some spinel was observed in the parent glass. As a result of the low Cr2O3 solubility in silicate melts, when its concentration is higher than 0.7–1.0 wt %, a part of this oxide spontaneously precipitates into “preliminary” spinel crystals during the melt cooling; the residual part eventually might participate in the formation of a “secondary” spinel phase at low temperatures. The “preliminary” spinel (labelled S1) that was formed in our glass was defined as a solid solution between MgO·Cr2O3 (ICSD171106) and FeO·Fe2O3 (ICSD158742).
In sample GC60-1, the formation of some pyroxene phase (labelled P1) was also identified. After 5 min crystallization at 730 °C, some increasing in its amount were observed. In the final glass-ceramics (see
Figure 3d), together with P1, a new pyroxene phase, one which practically becomes the main crystal phase in the material (labelled P2), is formed. According to the structural analysis P1 is characterized with a unit cell with V~428 × 10
6 pm
3, whereas in P2, V~422 × 10
6 pm
3. This difference is about 1.5%, and it clearly highlights a variation in the chemical compositions of both pyroxene phases. It can be assumed that some larger ions participate in the formation of the P1 phase. Additionally, some increase of the spinel phase is observed, which must be related to the formation of “secondary” magnetite spinel (labelled S2).
The results for the non-nucleated samples (GC0-1, GC0-5 and GC0-60) show no significant differences from the nucleated samples.
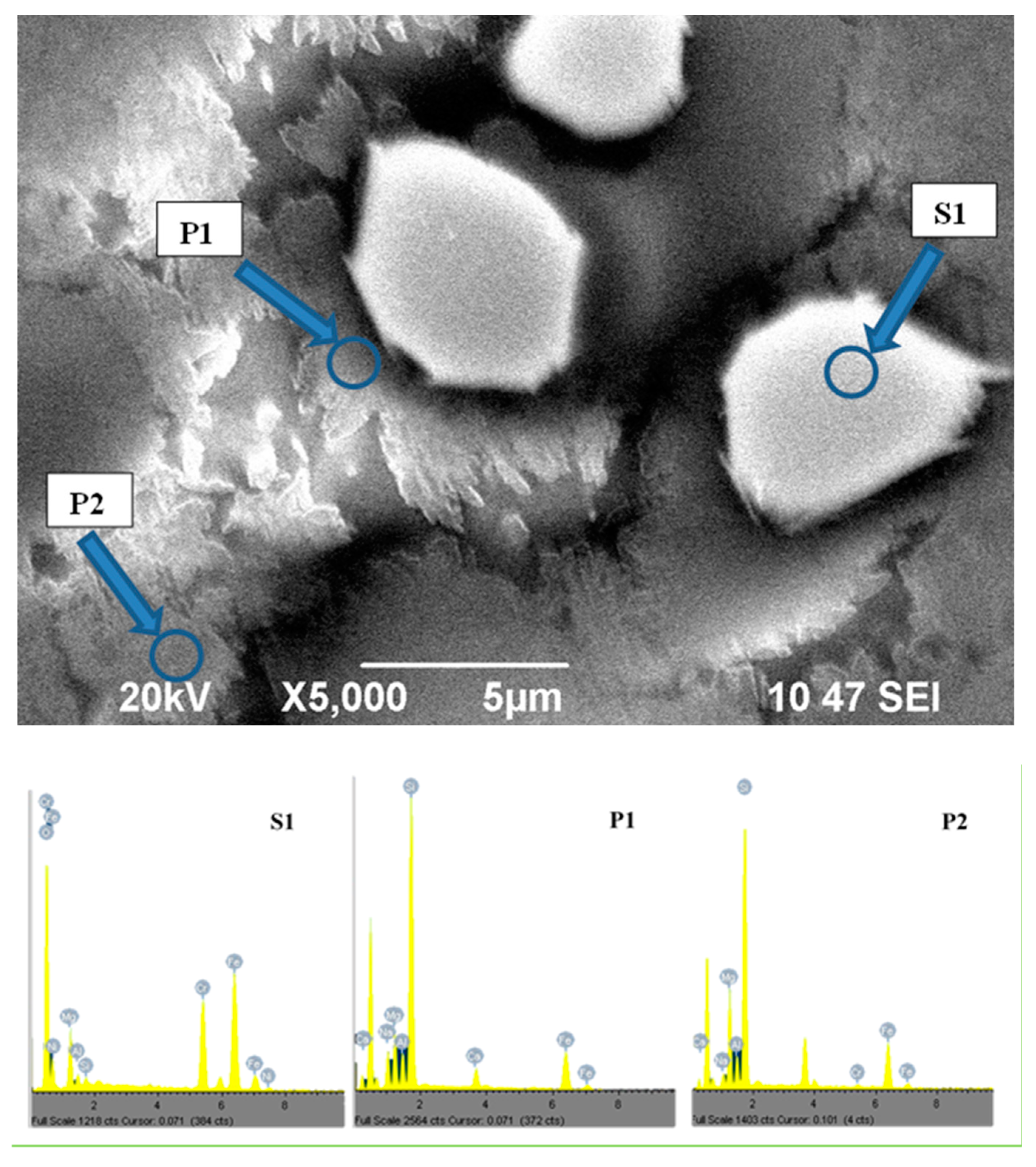
After 1 min at 730 °C, the phase formation is still in its initial stage and the SEM image, which is presented in
Figure 4a, practically shows only “preliminary” spinel crystals. Their size is between 3 and 10 μm, and the EDS analysis reports a quite constant chemical composition (mol %): 36 ± 2 iron oxides, 23 ± 2 Cr
2O
3 and 41 ± 3 MgO. These results confirm the XRD data for the formation of a solid solution between MgO·Cr
2O
3 and FeO·Fe
2O
3.
Since the molar percentage of MgO in the parent glass is higher than that of Cr2O3, it follows that the iron taking part in the formation of the “preliminary” spinel is predominantly in Fe3+ valence. This indicates that some increase of the Fe2+/Fe3+ ratio in the main glassy matrix can be expected, which must favor the subsequent formation of a “secondary” magnetite spinel phase. In addition, considering that the overall volume of the Cr-Mg-Fe spinel phase is at about 4–6%, it might also be concluded that the chromium oxide is mainly concentrated in the “preliminary” spinel crystals.
Figure 4b shows a photo at higher magnification, with a group of “preliminary” spinel crystals in GC60-1. It is evident that around these crystals is formed a “homogeneous” crystallization field with a thickness between 5 and 10 µm.
However, the main volume of the sample (i.e., at about 80–85%), where the Fe2O3 concentration is high, is characterized with a “non-homogeneous” structure due to the formation of tiny spherical particles enriched with iron. It is clear that these drops become finer and finer when moving away from the crystallization field.
Figure 5a,b elucidates areas free of “preliminary” spinel in samples GC0-1 and GC60-1, respectively. These SEM results demonstrate that without treatment at 650 °C, the size of iron-rich drops is in the range of 150–250 nm, while after the “nucleation step” it decreases to 50–70 nm and the number of drops significantly rises (from 15 ± 5 per µm
2 in GC0-1 to 50 ± 10 for GC60-1). This result clarifies that during the nucleation step binodal liquid–liquid separation occurs.
The liquid–liquid immiscibility was also confirmed by TEM observations. In
Figure 6a, a flake from the parent glass rapidly quenched in water, elucidating a homogeneous amorphous structure, is shown. On the contrary, in
Figure 6b, an image from sample GC60-1, where the structure is non-homogeneous and comparable to one obtained by SEM, is presented. In addition, the corresponding electron diffraction (SAED) pattern (shown in the inset) confirms the amorphous structure of GC60-1 and thus the assumption for initial liquid–liquid separation.
In samples GC0-5 and GC60-5, variations in the composition and the size of the “preliminary” spinel were not detected, which supports the hypothesis that this phase is generally formed during the melt’s cooling. In agreement with XRD results, the beginning of pyroxene growth on these spinel crystals was observed. It can be also supposed that at subsequent heat treatments, the iron-rich drops were transformed into tiny magnetite crystals, which than acted as nucleation agents for the pyroxene formation.
In sample GC0-60, presented in
Figure 7, together with the pyroxene crystals growing on the “preliminary” spinel, consistent crystallization (in the form of pyroxene spherulites with of 3–6 μm size) was also observed in the main matrix. In this figure, EDS spectra of the “preliminary” spinel and both pyroxene phases are also reported. The results demonstrate that the first pyroxene phase, growing in the diffusion field on the “preliminary” spinel, is characterized by higher concentrations of Ca
2+ and Na
+, while the main pyroxene phase was characterized by higher concentrations of Mg
2+. These results confirm the XRD data for both pyroxene phases and explain the differences in the volumes of their unit cells. In sample GC60-60, due to the effective “nucleation”, the size of both pyroxene crystals (P1 and P2) is significantly lower [
21]; in order to better highlight the structure of the final glass-ceramic, it was additionally studied by FESEM and EDS mapping of the elements Fe, Cr, Mg, Ca, Na and Si.
Figure 8 presents an image of a zone containing two “preliminary” spinel crystals and many 0.3–0.6 μm pyroxene P1 crystals growing on them in a preferential direction. The formation of tiny “secondary” spinel and a fine pyroxene P2 phase is also well distinguished in the circumference zone. It is also well seen that the sizes of the “secondary” spinel decrease with increased distance from the diffusion zone.
The EDS mapping highlights that the concentrations of Si, Na and Ca are higher in the diffusion field, while the ones of Fe and Mg are higher into the main matrix, which is in agreement with EDS data for both P1 and P2 pyroxenes from
Figure 7. It was also confirmed that the amount of Cr outside of the “preliminary” spinel is negligible, so the “secondary” spinel phase (S2) has a composition close to that of magnetite.
The main structure of sample GC60-60 (i.e., the part free from “preliminary” spinel), where P2 is the predominate crystal phase, is shown in
Figure 9. It highlights the high degree of crystallinity of the final glass-ceramic and shows that the size of formed pyroxenes having the typical prismatic attributes is about 200 nm. The additional analysis demonstrates that the number of these pyroxene crystals is 80 ± 15 per µm
2. This value is just slightly higher than that of one of the iron-rich drops formed in sample GC60-1, which confirms that the “secondary” spinel phase acts as nucleation agent for the main crystallization process.
Finally, TEM observation of the final glass-ceramic elucidates that the main P2 phase is composed of mono-crystals.
Figure 10 shows a single pyroxene crystal from sample GC60-60, which is embedded in an amorphous glass matrix with lower contrast. The SAED patterns and the HRTEM fringes reveal the single crystalline structure of the particle and its orientation along zone axis /11-4/. The phase identification highlights a monoclinic clinopyroxene with cell parameters a = 9.724, b = 8.875, c = 5.281 and β = 107.24 (PDF 85-1740), which is in an agreement with the XRD data.
4.3. Characteristics of the Final Glass-Ceramic
The main characteristics of the new material were evaluated by different techniques [
21]. Here these results are summarized in
Table 5, together with the corresponding data for Slagsitalls [
24,
25], which up to now have been the most-produced glass-ceramics in the world. Slagsitalls were developed in the former Soviet Union by Prof. Kitajgorodski only few years after the discovery of glass-ceramics by Prof. Stookey in “Corning”, New York, in the USA. Their production was mainly based on the use of blast furnace slag from the region of Donbas, Ukraine. In 1966 in the plants of Avtosteklo (Konstantinovka), a manufacturing line with daily capacity of 3000 m
2 was started, and by 1980 the entire volume of produced Slagsitall tiles surpassed 15,000,000 m
2.
The melting temperatures of the Slagsitalls batches, as well as those of various similar glass-ceramics, were at about 1500–1550 °C, while the nucleation and crystallization temperatures were at about 600–650 and 900–950 °C, respectively; the duration of the crystallization cycle was at about 3–4 h.
Notwithstanding the significantly lower melting and crystallization temperatures, as well as the shorter holding times for melting, nucleation and crystallization, the newly obtained iron-oxide-rich glass-ceramic is characterized by enhanced hardness and bending strength, both of which can be explained by the nano-structure of the material. At the same time, the values for compressive strength and Young’s modulus, which are high enough for a modern building material, are lower than the ones reported for the Slagsitalls. This is a consequence of the non-homogeneous structure of new iron-rich materials due to the precipitation of “preliminary” spinel. Finally, due to the formation of crystal phases as iron rich pyroxenes and spinel, the density of our glass-ceramic is slightly higher, while the coefficient of thermal expansion is moderate, even a little lower than the one reported for the Slagsitalls.
Finally, the TCLP tests of the parent glass and the final glass-ceramics, reported in
Table 2, demonstrate that the new material is completely inert. In fact, the measured concentrations of heavy metals are two to four orders of magnitude lower than those of the corresponding values for the initial residues.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}