1. Introduction
In the task of comparison and analysis of biological sequences, choosing a type of DNA/protein representation is an important step. The usual representation of the primary structure of DNA is a string of four letters: A (adenine); G (guanine); C (cytosine); and T (thymine). This expression is called a letter sequence representation (LSR) or a DNA primary sequence. Similarly, a protein primary sequence is usually expressed in terms of a series of 20 letters, which denote 20 different amino acids. The sequence encodes information of the corresponding structure and function in a living organism. However, it is difficult to obtain the information from the representation of a primary sequence directly. Therefore, various sequence representation techniques have been developed for encoding bio-sequences and extracting the hidden information.
Graphical representation of DNA is a useful tool for visualizing and analyzing DNA sequences. By using the tool, one can obtain a route to condense the information coded by DNA primary sequences into a set of invariants [
1,
2]. Early attempts towards graphical representations of DNA were made by Hamori and Ruskin in 1983 [
3], Hamori in 1985 [
4], and Gates in 1985 [
5]. Afterwards, more graphical representations of DNA sequences were well developed by researchers [
1,
2,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15]. In comparison with DNA, graphical representations of proteins emerged only very recently [
2,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27]. As a matter of fact, most of the graphical representations of DNA involve some degree of arbitrariness, such as the selection of directions to be assigned to individual bases. For a string like DNA sequence over an alphabet with size 4, there are
possible ways of assigning 4 directions to 4 nucleic acid bases. If these methods are directly extended to protein sequences, the corresponding figure is
. It is impracticable to represent one protein sequence by such an enormous number of graphs. This is probably the most important reason why protein graphical representations have not been advanced [
19,
23]. It is found that reducing the alphabet or fixing the directions assigned to amino acid residues plays an important role in addressing this problem. For details, we refer to some recent publications [
2,
16,
21,
23,
24,
28].
Matrix representation of a biological sequence is another powerful tool for characterization and comparison of sequences. These matrices include: The frequency matrix; Euclidean-distance matrix (
ED); graph theoretical distance matrix (
GD); line distance matrix (
LD); quotient matrix (
D/D,
M/M,
L/L); and their “higher order” matrices [
1,
2,
12,
13,
20,
21,
27,
29,
30]. Among them,
ED,
GD,
L/L, etc., are derived from a graphical representation. For example,
L/L is a symmetric matrix whose diagonal entries are zero, while other entries are defined as the quotient of the Euclidean distance between two points of the graph and the sum of geometrical lengths of edges between the two points. Once the matrix is given, some of matrix invariants can be used as descriptors of the sequence. Eigenvalues of a matrix are one of the best-known matrix invariants [
31]. In fact, two graphs are isomorphic if and only if their adjacency matrices are similar. It is of interest to note that similar matrices have the same eigenvalues. Among all the eigenvalues, the leading eigenvalue often plays a special role and has been widely used in the field of biological science and chemistry. However, a problem we must face is that the calculation of the eigenvalue will become more and more difficult with the order of the matrix large. ALE-index is an alternative invariant we proposed in 2005 [
32]. The ALE-index can be viewed as an Approximation of the Leading Eigenvalue (ALE) of the corresponding matrix (it is just in this sense that it is called ‘ALE’-index), while it is much simpler for calculation than the latter. Therefore, it may be more economical to adopt the ALE-index when one is interested only in the leading eigenvalue.
The third method for formulating a protein sequence is the pseudo amino acid composition (PseAAC), with the advantage of avoiding loss of the sequence-order information. Ever since the concept of PseAAC [
33,
34] or Chou’s PseAAC [
35,
36] was proposed, it has rapidly penetrated into nearly all fields of computational proteomics (see a long list papers cited in [
36,
37]). Stimulated by the great successes of PseAAC in dealing with protein/peptide sequences, the concept of PseAAC has been extended [
38,
39,
40,
41,
42] to cover DNA/RNA sequences as well via the form of PseKNC (pseudo K-tuple nucleotide composition) [
43,
44], which has been proven very useful in studying many important genome analysis problems, as summarized in a recent review paper [
45]. Also, because the concept of PseAAC has been increasingly and widely used in both computational proteomics and genomics, a very powerful web-server called “Pse-in-One” [
46] was established that can be used to generate the pseudo components for both protein/peptide and DNA/RNA sequences.
In this paper, we modify the method of Chou’s PseAAC and propose a novel approach for numerically characterizing a protein sequence. We characterize a protein sequence by a
dimensional vector, whose first 20 components are the occurrence frequencies of 20 native amino acids, while the last
components are based on a six-letter sequence derived from the protein primary sequence. The former is used to reflect the effect of the amino acid composition, and the latter is used to reflect the effect of sequence order and property of the residues. It is well known that a sequence naturally contains two pieces of information: the elements of the sequence; and the orders of the elements. Any methodologies based on the amino acid composition alone are worthy of further investigation. However, as pointed out by Chou [
33,
34], it is not feasible to completely include all sequence order patterns. It was stirring to see that Chou creatively developed an approach as mentioned above to extract the important feature beyond amino acid composition. Our scheme is similar to, but different from, that of Chou. Experiments about phylogenetic analysis on two datasets and identification of DNA-binding proteins illustrate the utility of the proposed method.
2. Methods
A protein sequence can be viewed as a string of 20 amino acids. Without loss of generality, by the numerical indices 1,2,…,20, we represent the 20 native amino acids according to the alphabetical order of their single-letter codes: A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W and Y. Then the frequencies of appearance of the 20 amino acids in a protein sequence are often used to construct a vector
This is the conventional amino acid composition. The advantage of such a vector representation is that it is easy in statistical treatment, but it cannot reflect the effect regarding sequence order and property. In what follows, we will take this effect into account through a set of elements in addition to the 20 components.
Hydrophobicity, isoelectric point (pI), and relative distance (RD) are three important physicochemical properties of the 20 native amino acids. Here RD can be viewed as an integration of the information on three side chain properties: composition; polarity; and molecular volume—where composition is defined as the atomic weight ratio of hetero (noncarbon) elements in end groups or rings to carbons in the side chain (for details, see [
47]). Listed in
Table 1 are the original numerical values for hydrophobicity, pI and RD. As can be seen from
Table 1, the values of
(Hydrophobicity) is in the range [−2.53~1.38], and the values of
(isoelectric point) are in the range of 2.97~10.76, while
(relative distance) varies between 1469 and 3355. Therefore, the normalization of these values is needed. Here we normalize them by the formulary below:
Clearly, the normalized values for properties of the 20 native amino acids are in the interval [0,1]. The corresponding values are listed in
Table 2. The last row in this table gives the average values.
For each amino acid (AA), we associate it with a triple (
t(1),
t(2),
t(3)), where
All the amino acids with a same triple form a group. In this way, the 20 native amino acids can be classified into 6 groups:
GI = {A, Y, V, M, L, I},
GII = {C, W, G, F},
GIII = {D, S, N},
GIV = {E, Q},
GV = {H, T, R, K},
GVI = {P}.
For each group, the first amino acid is selected to be the representative. That is, A, C, D, E, H and P are used to stand for the six groups, respectively. The value of the property of a group is defined as the average value of the property of amino acids belonging to the group. Listed in
Table 3 are the corresponding values of the six groups.
At the same time, a protein primary sequence can be reduced into a six-letter sequence by replacing each element in the protein sequence with its representative letter. Suppose
is a given six-letter sequence, we inspect it by stepping one element at a time. For the step
k (
), a 3-D space point
can be constructed as follows:
where
. When
k runs from 1 to
L, we get
L points
. Connecting these points one by one sequentially with straight lines, a three-dimensional curve can be drawn. One can further associate the graph with some structural matrices. Here we adopt the
L/L matrix and denote it by
M, whose (
i,j)-entry is defined as follows:
where
is the Euclidean distance between points
and
. It is not difficult to see that
tM is a (0,1) matrix; here
tM stands for the product of Hadmammard multiplication of the matrix
M by itself
t-times. In this paper, we call the limit matrix as a generalized adjacency matrix (GAM) generated by points
, and denote it by
MG. Obviously,
if and only if
and
lie on a straight line in the graph.
As mentioned above, once a symmetric matrix is given, one can calculate its ALE-index by the following formula:
where
L is the order of the matrix,
and
are the
m1- and
F-norms of a matrix, respectively. In order to reduce variations caused by comparison of matrices with different sizes, we consider instead of
a normalized ALE-index
.
In addition, following the similar procedures in capturing the sequence-order information of a protein [
33,
34], for the six-letter sequence
, we extract a set of new order-correlated factors as defined below:
where
is called the
k-th tier correlation factor,
represents the coupling mode function as given by
Factor
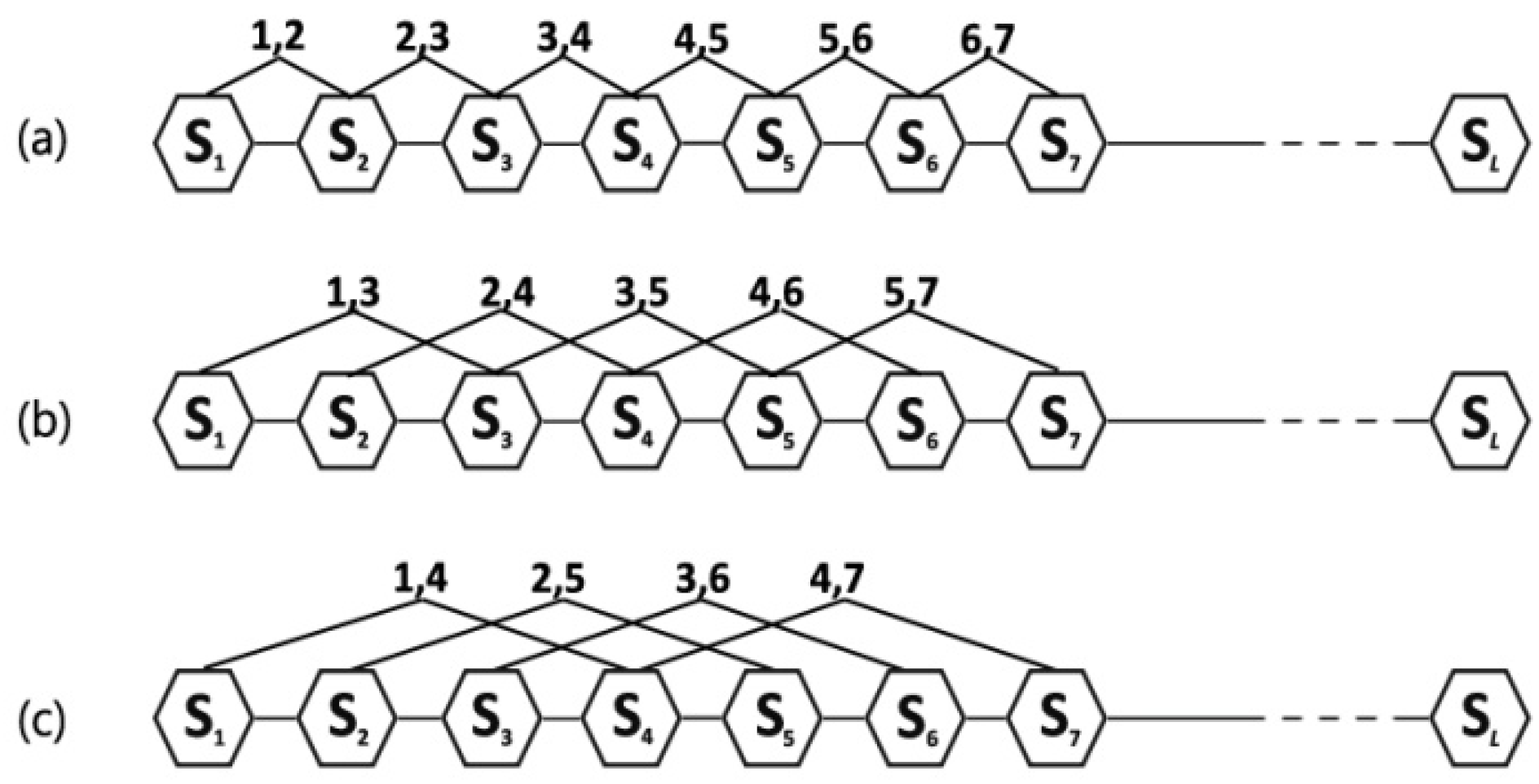
reflects the coupling mode between the most contiguous elements along a six-letter sequence (
Figure 1a);
reflects the coupling mode between the second-most contiguous (
Figure 1b);
reflects the coupling mode between the third-most contiguous (
Figure 1c), and so on.
is the highest rank of the coupling mode.
Consequently, a protein sequence can be characterized by a
dimensional vector
V:
where
Here and are weight factors. It is easy to see that the first 20 components reflect the effect of the amino acid composition, whereas the last components reflect the effect of sequence order and property of the residues. For convenience, a set of such components as formulated by Equations (8) and (9) is called the generalized pseudoamino acid composition of a protein sequence, and denoted by G-PseAAC.







{kind=link}
{kind=link}
{kind=link}
{kind=link}