The Electronic, Magnetic, Half-Metallic and Mechanical Properties of the Equiatomic Quaternary Heusler Compounds FeRhCrSi and FePdCrSi: A First-Principles Study


Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
3. Results
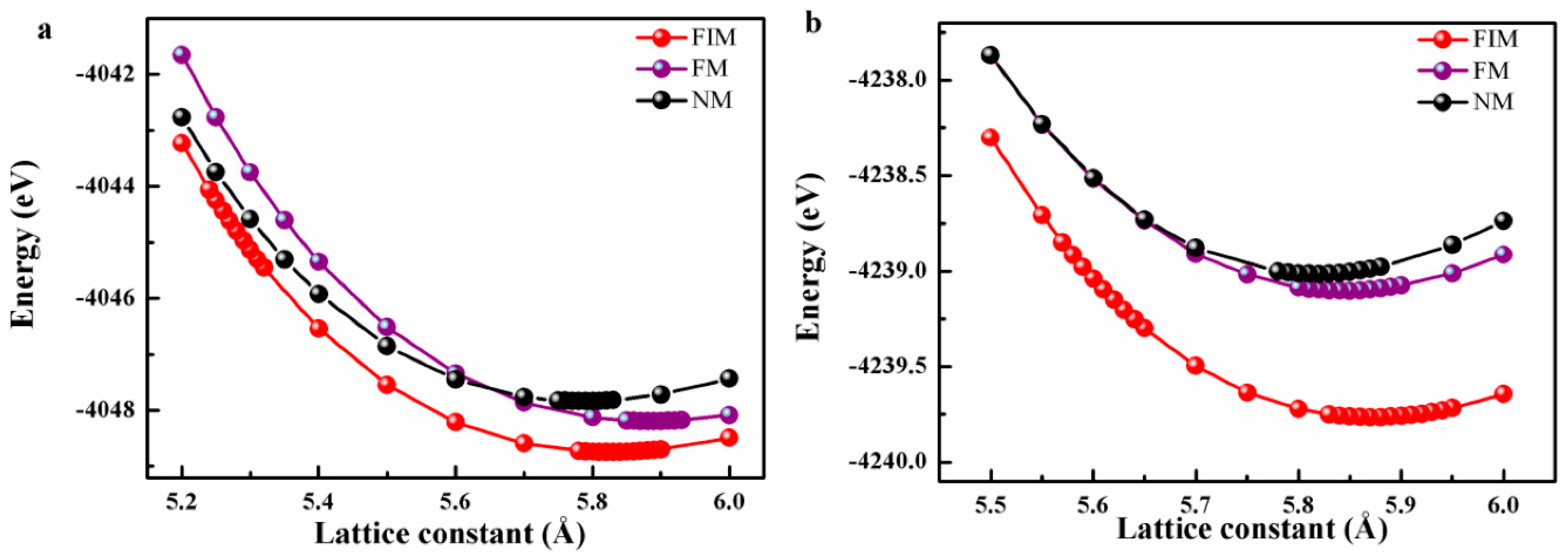
3.1. Total Energy and Structural Stability
3.2. Electronic, Magnetic, and Half-Metallic Properties
3.3. Structural Stability
3.4. Mechanical Properties
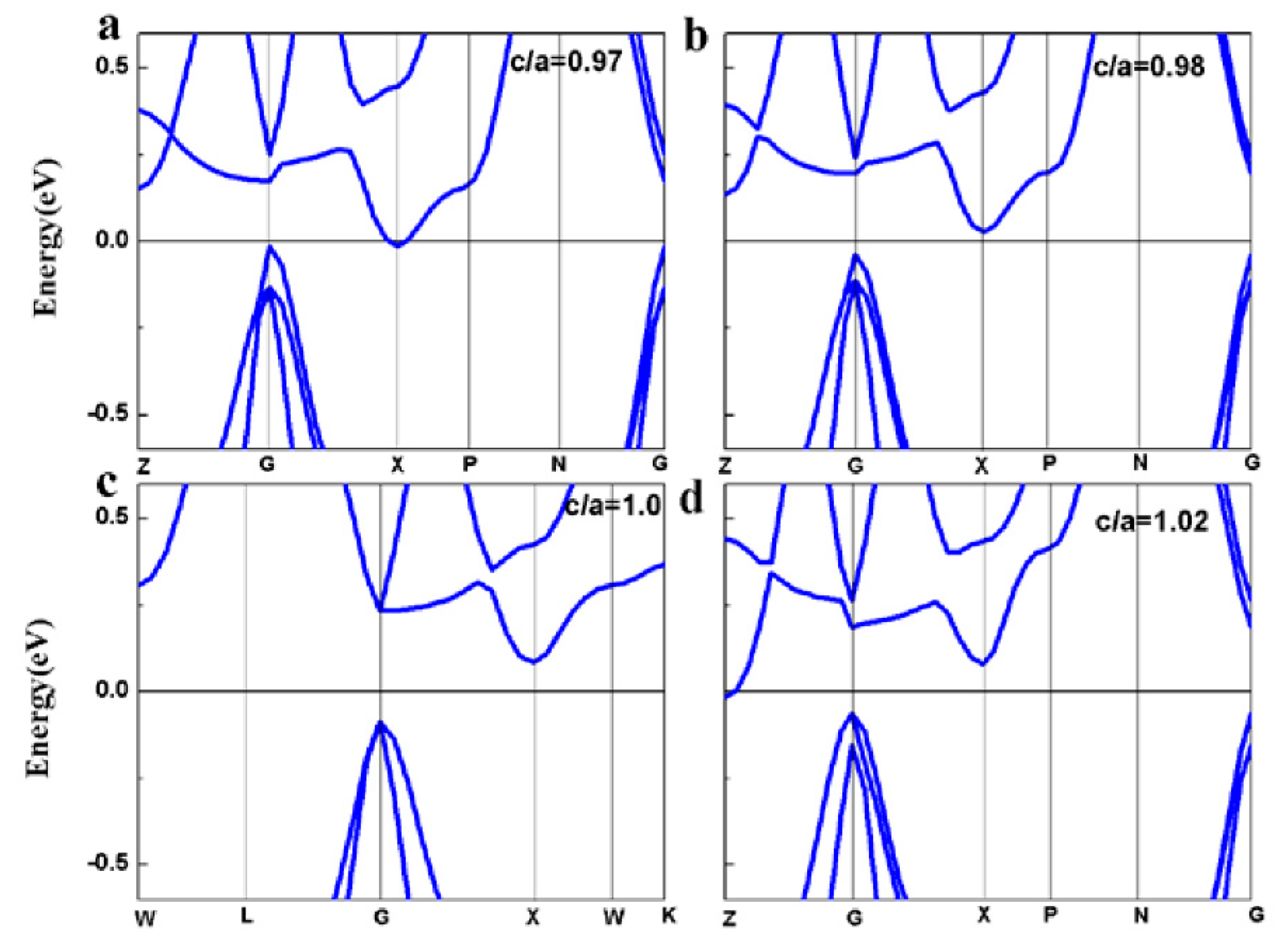
3.5. Strain Effect on the Electronic and Magnetic Properties
4. Conclusions
- (1)
- FeRhCrSi and FePdCrSi compounds are two new ferrimagnetic HMs with a wide half-metallic band gap of 0.336 eV and 0.177 eV, respectively.
- (2)
- The half-metallicity of the FeRhCrSi and FePdCrSi compounds are very robust to the hydrostatic strain or tetragonal distortion. Especially for FeRhCrSi, the half-metallicity can be kept in a wide lattice constant range (5.28 Å–5.85 Å) under hydrostatic strain and a c/a ratios range (0.98–1.08) under tetragonal distortion, respectively.
- (3)
- The total magnetic moment of FeRhCrSi and FePdCrSi compounds are 3 μB and 4 μB, respectively, which obey the Slater–Pauling rule: Mt = Zt-24. The main contributor of the total magnetic moments are both Cr atom for FeRhCrSi and FePdCrSi.
- (4)
- The large negative values of the calculated formation energy and cohesion energy show the direct evidence of the chemical and thermal stability for FeRhCrSi and FePdCrSi compounds. This indicates that they are likely to be synthesized in the experiment.
- (5)
- The elastic constants and the various moduli indicate the mechanical stability of FeRhCrSi and FePdCrSi compounds.
Author Contributions
Funding
Conflicts of Interest
References
- De Groot, R.A.; Mueller, F.M.; van Engen, P.G.; Buschow, K.H.J. New class of materials: half-metallic ferromagnets. Phys. Rev. Lett. 1983, 50, 2024. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, Z.; Jin, Y.; Wu, Y.; Dai, X.; Liu, G. Magneto-electronic properties and tetragonal deformation of rare-earth-element-based quaternary Heusler half-metals: A first-principles prediction. J. Alloys Compd. 2018, 734, 329–341. [Google Scholar] [CrossRef]
- Sun, Q.; Kioussis, N. Prediction of manganese trihalides as two-dimensional Dirac half-metals. Phys. Rev. B 2018, 97, 094408. [Google Scholar] [CrossRef]
- Davatolhagh, S.; Dehghan, A. Dirac-like half-metallicity of d0 − d half-Heusler alloys. Phys. C Supercond. Appl. 2018, 552, 53–56. [Google Scholar] [CrossRef]
- Han, Y.; Wang, X. First-Principles Investigation of Half-Metallic Ferromagnetism of a New 1:1:1:1 Type Quaternary Heusler Compound YRhTiSi. J. Supercond. Novel Magn. 2018. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.T.; Rozale, H.; Gao, Y.C.; Wang, L.Y.; Chen, X.B. Electronic structures, magnetic properties and half-metallicity in Heusler alloys Zr2IrZ (Zá = áAl, Ga, In). Curr. Appl. Phys. 2015, 15, 1117–1123. [Google Scholar] [CrossRef]
- Yu, W.; Zhu, Z.; Niu, C.Y.; Li, C.; Cho, J.H.; Jia, Y. Dilute Magnetic Semiconductor and Half-Metal Behaviors in 3 d Transition-Metal Doped Black and Blue Phosphorenes: A First-Principles Study. Nanoscale research letters. Nanoscale Res. Lett. 2016, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Galanakis, I.; Mavropoulos, P. Zinc-blende compounds of transition elements with N, P, As, Sb, S, Se, and Te as half-metallic systems. Phys. Rev. B 2003, 67, 104417. [Google Scholar] [CrossRef]
- Wurmehl, S.; Fecher, G.H.; Kandpal, H.C.; Ksenofontov, V.; Felser, C.; Lin, H.J. Investigation of Co 2 Fe Si: The Heusler compound with highest Curie temperature and magnetic moment. Appl. Phys. Lett. 2005, 88, 032503. [Google Scholar] [CrossRef]
- Kanomata, T.; Kyuji, S.; Nashima, O.; Ono, F.; Kaneko, T.; Endo, S. The Curie temperature in Heusler alloys Ni2MnZ (Z = Ga, Sn and Sb) under high pressure. J. Alloys Compd. 2012, 518, 19–21. [Google Scholar] [CrossRef]
- Campbell, C.C.M. Hyperfine field systematics in Heusler alloys. J. Phys. F: Met. Phys. 1975, 5, 1931. [Google Scholar] [CrossRef]
- Peng, H.; Perdew, J.P. Rehabilitation of the Perdew-Burke-Ernzerhof generalized gradient approximation for layered materials. Phys. Rev. B 2017, 95, 081105. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zhao, Y.; Li, Z.; Chen, H.; Tao, X.; Ouyang, Y. The effect of Al content on the structural, mechanical, and thermal properties of B2-FeAl and D03-Fe3Al from atomistic study. Mater. Res. Express 2018, 5, 026512. [Google Scholar] [CrossRef]
- Jamer, M.E.; Wang, Y.J.; Stephen, G.M.; McDonald, I.J.; Grutter, A.J.; Sterbinsky, G.E.; Arena, D.A.; Borchers, J.A.; Kirby, B.J.; Lewis, L.H.; et al. Compensated Ferrimagnetism in the Zero-Moment Heusler Alloy Mn 3 Al. Phys. Rev. Appl. 2017, 7, 064036. [Google Scholar] [CrossRef]
- Helmholdt, R.B.; de Groot, R.A.; Mueller, F.M.; van Engen, P.G.; Buschow, K.H.J. Magnetic and crystallographic properties of several C1b type Heusler compounds. J. Magn. Magn. Mater. 1984, 43, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Bainsla, L.; Suresh, K.G. Equiatomic quaternary Heusler alloys: A material perspective for spintronic applications. Appl. Phys. Rev. 2016, 3, 031101. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Liu, G.; Fecher, G.H.; Felser, C.; Li, Y.; Liu, H. New quarternary half metallic material CoFeMnSi. J. Appl. Phys. 2009, 105, 07E901. [Google Scholar] [CrossRef]
- Klaer, P.; Balke, B.; Alijani, V.; Winterlik, J.; Fecher, G.H.; Felser, C.; Elmers, H.J. Element-specific magnetic moments and spin-resolved density of states in CoFeMn Z (Z = Al, Ga; Si, Ge). Phys. Rev. B 2011, 84, 144413. [Google Scholar] [CrossRef]
- Alijani, V.; Ouardi, S.; Fecher, G.H.; Winterlik, J.; Naghavi, S.S.; Kozina, X.; Stryganyuk, G.; Felser, C.; Ikenaga, E.; Yamashita, Y.; et al. Electronic, structural, and magnetic properties of the half-metallic ferromagnetic quaternary Heusler compounds CoFeMn Z (Z = Al, Ga, Si, Ge). Phys. Rev. B 2011, 84, 224416. [Google Scholar] [CrossRef]
- Alijani, V.; Winterlik, J.; Fecher, G.H.; Naghavi, S.S.; Felser, C. Quaternary half-metallic Heusler ferromagnets for spintronics applications. Phys. Rev. B 2011, 83, 184428. [Google Scholar] [CrossRef]
- Gao, G.Y.; Hu, L.; Yao, K.L.; Luo, B.; Liu, N. Large half-metallic gaps in the quaternary Heusler alloys CoFeCrZ (Z = Al, Si, Ga, Ge): A first-principles study. J. Alloys Compd. 2013, 551, 539–543. [Google Scholar] [CrossRef]
- Karimian, N.; Ahmadian, F. Electronic structure and half-metallicity of new quaternary Heusler alloys NiFeTiZ (Z = Si, P, Ge, and As). Solid State Commun. 2015, 223, 60–66. [Google Scholar] [CrossRef]
- Berri, S.; Ibrir, M.; Maouche, D.; Attallah, M. Robust half-metallic ferromagnet of quaternary Heusler compounds ZrCoTiZ (Z = Si, Ge, Ga and Al). Comput. Condens. Matter 2014, 1, 26–31. [Google Scholar] [CrossRef]
- Wang, X.T.; Cheng, Z.X.; Guo, R.K.; Wang, J.L.; Rozale, H.; Wang, L.Y.; Yu, Z.Y.; Liu, G.D. First-principles study of new quaternary Heusler compounds without 3d transition metal elements: ZrRhHfZ (Z= Al, Ga, In). Mater. Chem. Phys. 2017, 193, 99–108. [Google Scholar] [CrossRef]
- Wang, X.T.; Khachai, H.; Khenata, R.; Yuan, H.K.; Wang, L.Y.; Wang, W.H.; Bouhemadou, A.; Hao, L.Y.; Dai, X.F.; Guo, R.K.; et al. Structural, electronic, magnetic, half-metallic, mechanical, and thermodynamic properties of the quaternary Heusler compound FeCrRuSi: A first-principles study. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Liu, G.; Wang, X.; Rozale, H.; Wang, L.; Khenata, R.; Wu, Z.; Dai, X. First-principles study on quaternary Heusler compounds ZrFeVZ (Z = Al, Ga, In) with large spin-flip gap. RSC Adv. 2016, 6, 109394–109400. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J. First-principles simulation: ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Zeitschrift für Kristallographie-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Stephens, P.J. Hartree-Fock and density functional theory ab initio calculation of optical rotation using GIAOs: Basis set dependence. J. Phys. Chem. A 2000, 104, 1039. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671. [Google Scholar] [CrossRef]
- Steinmann, S.N.; Csonka, G.; Corminboeuf, C. Unified Inter- and Intramolecular Dispersion Correction Formula for Generalized Gradient Approximation Density Functional Theory. J. Chem. Theory Comput. 2009, 5, 2950. [Google Scholar] [CrossRef] [PubMed]
- Umari, P.; Pasquarello, A. Polarizability and dielectric constant in density-functional supercell calculations with discrete k-point samplings. Phys. Rev. B 2003, 68, 085114. [Google Scholar] [CrossRef]
- Rath, J.; Freeman, A.J. Generalized magnetic susceptibilities in metals: Application of the analytic tetrahedron linear energy method to Sc. Phys. Rev. B 1975, 11, 2109. [Google Scholar] [CrossRef]
- Galanakis, I.; Dederichs, P.H.; Papanikolaou, N. Slater-Pauling behavior and origin of the half-metallicity of the full-Heusler alloys. Phys. Rev. B 2002, 66, 174429. [Google Scholar] [CrossRef]
- Galanakis, I. Slater-Pauling Behavior in Half-Metallic Magnets. J. Surf. Interface. Mater. 2014, 2, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Fecher, G.H.; Kandpal, H.C.; Wurmehl, S.; Felser, C.; Schönhense, G. Slater-Pauling rule and Curie temperature of Co2Co2-based Heusler compounds. J. Appl. Phys. 2006, 99, 08J106. [Google Scholar] [CrossRef]
- Srivastava, G.P.; Weaire, D. The theory of the cohesive energies of solids. Adv. Phys. 1987, 36, 463. [Google Scholar] [CrossRef]
- Huang, K.; Born, M. Clarendon. In Dynamical Theory of Crystal Lattices; Clarendon Press: Oxford, UK, 1954; p. 420. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Fe | Rh/Pd | Cr | Si |
|---|---|---|---|---|
| Type 1 | 4a | 4c | 4b | 4d |
| Type 2 | 4b | 4c | 4a | 4d |
| Type 3 | 4a | 4b | 4c | 4d |
| Compounds | a0 | Mtot | MFe | MRh/Pd | MCr | MSi | Ef | Ec |
|---|---|---|---|---|---|---|---|---|
| FeRhCrSi | 5.82 | 3.00 | −0.26 | 0.22 | 3.10 | −0.06 | 3.12 | −20.41 |
| FePdCrSi | 5.87 | 4.00 | 0.78 | −0.14 | 3.44 | −0.08 | −1.97 | −18.66 |
| Compounds | C11 | C12 | C44 | B | G | E | B/G | A |
|---|---|---|---|---|---|---|---|---|
| FeRhCrSi | 294.7 | 112.9 | 106.6 | 173.5 | 100.0 | 251.7 | 1.74 | 1.17 |
| FePdCrSi | 179.4 | 121.5 | 75.1 | 140.8 | 45.9 | 124.2 | 3.07 | 2.59 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, L.; Ma, J.; Yang, Y.; Lin, T.; Wang, L. The Electronic, Magnetic, Half-Metallic and Mechanical Properties of the Equiatomic Quaternary Heusler Compounds FeRhCrSi and FePdCrSi: A First-Principles Study. Appl. Sci. 2018, 8, 2370. https://doi.org/10.3390/app8122370
Feng L, Ma J, Yang Y, Lin T, Wang L. The Electronic, Magnetic, Half-Metallic and Mechanical Properties of the Equiatomic Quaternary Heusler Compounds FeRhCrSi and FePdCrSi: A First-Principles Study. Applied Sciences. 2018; 8(12):2370. https://doi.org/10.3390/app8122370
Chicago/Turabian StyleFeng, Liefeng, Jiannan Ma, Yue Yang, Tingting Lin, and Liying Wang. 2018. "The Electronic, Magnetic, Half-Metallic and Mechanical Properties of the Equiatomic Quaternary Heusler Compounds FeRhCrSi and FePdCrSi: A First-Principles Study" Applied Sciences 8, no. 12: 2370. https://doi.org/10.3390/app8122370
APA StyleFeng, L., Ma, J., Yang, Y., Lin, T., & Wang, L. (2018). The Electronic, Magnetic, Half-Metallic and Mechanical Properties of the Equiatomic Quaternary Heusler Compounds FeRhCrSi and FePdCrSi: A First-Principles Study. Applied Sciences, 8(12), 2370. https://doi.org/10.3390/app8122370