New Insights into Potocki-Shaffer Syndrome: Report of Two Novel Cases and Literature Review

 , ,
, ,  , , and
, , and 
Abstract
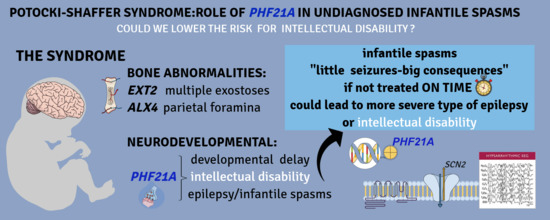
:
1. Introduction
2. Materials and Methods
3. Results
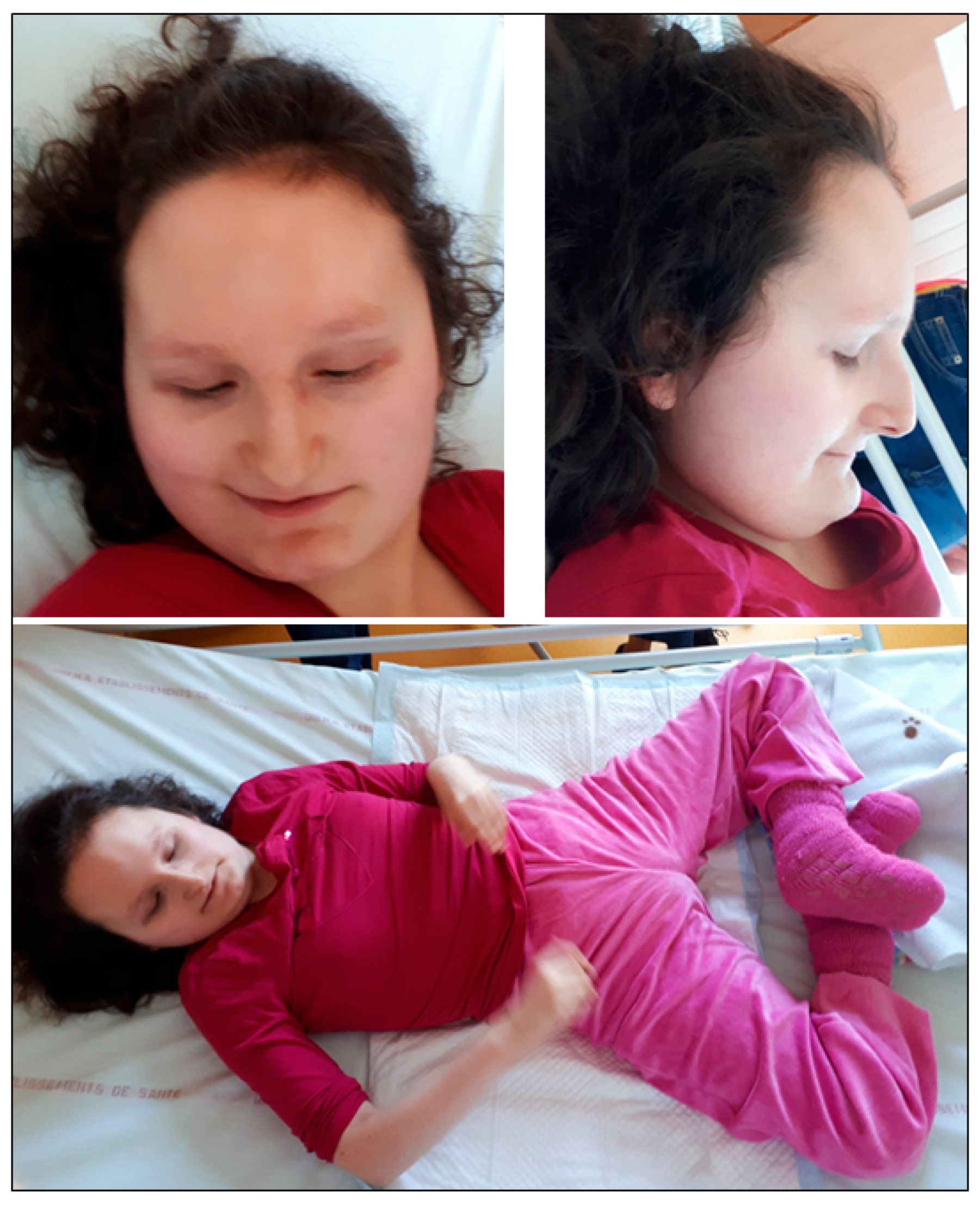
3.1. Case Report 1 (Decipher 286390)
3.2. Case Report 2 (Decipher 415213)
3.3. Review of Reported PSS Cases
4. Discussion
5. Conclusions
- -
- a detailed clinical examination of neonates, particularly focused on getting a full neurological assessment
- -
- a complete video-EEG recording
- -
- a magnetic resonance (MR) study of the brain
- Decipher: http://decipher.sanger.ac.ukWorkings
- OMIM, http://www.omim.org/
- UCSC Genome Browser, https://genome.ucsc.edu/
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural Variation in the Human Genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global Variation in Copy Number in the Human Genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuzun, E.; Sharp, A.J.; Bailey, J.A.; Kaul, R.; Morrison, V.A.; Pertz, L.M.; Haugen, E.; Hayden, H.; Albertson, D.; Pinkel, D.; et al. Fine-Scale Structural Variation of the Human Genome. Nat. Genet. 2005, 37, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Zhang, F.; Lupski, J.R. Genomic Disorders: A Window into Human Gene and Genome Evolution. Proc. Natl. Acad. Sci. USA 2010, 107, 1765–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankiewicz, P.; Inoue, K.; Bi, W.; Walz, K.; Park, S.S.; Kurotaki, N.; Shaw, C.J.; Fonseca, P.; Yan, J.; Lee, J.A.; et al. Genomic Disorders: Genome Architecture Results in Susceptibility to DNA Rearrangements Causing Common Human Traits. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2003; Volume 68, pp. 445–454. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Shaw, C.J.; Dapper, J.D.; Wakui, K.; Shaffer, L.G.; Withers, M.; Elizondo, L.; Park, S.S.; Lupski, J.R. Genome Architecture Catalyzes Nonrecurrent Chromosomal Rearrangements. Am. J. Hum. Genet. 2003, 72, 1101–1116. [Google Scholar] [CrossRef] [Green Version]
- Hastings, P.J.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of Change in Gene Copy Number. Nat. Rev. Genet. 2009, 10, 551–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pober, B.R. Williams-Beuren Syndrome. N. Engl. J. Med. 2010, 362, 239–252. [Google Scholar] [CrossRef]
- Boone, P.M.; Bacino, C.A.; Shaw, C.A.; Eng, P.A.; Hixson, P.M.; Pursley, A.N.; Kang, S.H.L.; Yang, Y.; Wiszniewska, J.; Nowakowska, B.A.; et al. Detection of Clinically Relevant Exonic Copy-Number Changes by Array CGH. Hum. Mutat. 2010, 31, 1326–1342. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy Number Variation in Human Health, Disease, and Evolution. Annu. Rev. Genom. Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Potocki, L.; Shaffer, L.G. Interstitial Deletion of 11(P11.2p12): A Newly Described Contiguous Gene Deletion Syndrome Involving the Gene for Hereditary Multiple Exostoses (EXT2). Am. J. Med. Genet. 1996, 62, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, L.G.; Hecht, J.T.; Ledbetter, D.H.; Greenberg, F. Familial Interstitial Deletion 11(P11.12p12) Associated with Parietal Foramina, Brachymicrocephaly, and Mental Retardation. Am. J. Med. Genet. 1993, 45, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Swarr, D.T.; Bloom, D.; Lewis, R.A.; Elenberg, E.; Friedman, E.M.; Glotzbach, C.; Wissman, S.D.; Shaffer, L.G.; Potocki, L. Potocki-Shaffer Syndrome: Comprehensive Clinical Assessment, Review of the Literature, and Proposals for Medical Management. Am. J. Med. Genet. A 2010, 152A, 565–572. [Google Scholar] [CrossRef]
- Brémond-Gignac, D.; Crolla, J.A.; Guichet, A.; Bonneau, D.; Taine, L.; Lacombe, D.; Baumann, C.; Benzacken, B.; Verloes, A. Combination of WAGR and Potocki-Schaffer Contiguous Deletion Syndromes in a Patient with an 11p11.2-P14 Deletion. Eur. J. Hum. Genet. 2005, 13, 409–413. [Google Scholar] [CrossRef]
- McGaughran, J.M.; Ward, H.B.; Evans, D.G.R. WAGR Syndrome and Multiple Exostoses in a Patient with Del (11) (P11.2p14.2). J. Med. Genet. 1995, 32, 823–824. [Google Scholar] [CrossRef] [Green Version]
- Wakui, K.; Gregato, G.; Ballif, B.C.; Glotzbach, C.D.; Bailey, K.A.; Kuo, P.L.; Sue, W.C.; Sheffield, L.J.; Irons, M.; Gomez, E.G.; et al. Construction of a Natural Panel of 11p11.2 Deletions and Further Delineation of the Critical Region Involved in Potocki-Shaffer Syndrome. Eur. J. Hum. Genet. 2005, 13, 528–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.O.; Howarth, R.J.; Williamson, K.A.; van Heyningen, V.; Beal, S.J.; Crolla, J.A. Genetic Analysis of Chromosome 11p13 and the PAX6 Gene in a Series of 125 Cases Referred with Aniridia. Am. J. Med. Genet. Part A 2008, 146, 558–569. [Google Scholar] [CrossRef]
- Miller-Hodges, E. Clinical Aspects of WT1 and the Kidney. Methods Mol. Biol. 2016, 1467, 15–21. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Brioude, F.; Toutain, A.; Giabicani, E.; Cottereau, E.; Cormier-Daire, V.; Netchine, I. Overgrowth Syndromes—Clinical and Molecular Aspects and Tumour Risk. Nat. Rev. Endocrinol. 2019, 15, 299–311. [Google Scholar] [CrossRef]
- Kim, H.G.; Kim, H.T.; Leach, N.T.; Lan, F.; Ullmann, R.; Silahtaroglu, A.; Kurth, I.; Nowka, A.; Seong, I.S.; Shen, Y.; et al. Translocations Disrupting PHF21A in the Potocki-Shaffer-Syndrome Region Are Associated with Intellectual Disability and Craniofacial Anomalies. Am. J. Hum. Genet. 2012, 91, 56–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeike, B.F.M.; Wuyts, W. Proximal Chromosome 11p Contiguous Gene Deletion Syndrome Phenotype: Case Report and Review of the Literature. Clin. Neuropathol. 2007, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.; Wakui, K.; Sue, W.C.; Su, M.H.; Shaffer, L.G.; Kuo, P.L. Interstitial Deletion 11(P11.12p11.2) and Analphoid Marker Formation Results in Inherited Potocki-Shaffer Syndrome. Am. J. Med. Genet. 2005, 133A, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, W.; Waeber, G.; Meinecke, P.; Schüler, H.; Goecke, T.O.; Van Hul, W.; Bartsch, O. Proximal 11p Deletion Syndrome (P11pDS): Additional Evaluation of the Clinical and Molecular Aspects. Eur. J. Hum. Genet. 2004, 12, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Chien, W.H.; Sue, W.C.; Kuo, P.L.; Su, M.H.; Lin, C.L. Potocki-Shaffer Syndrome: Report of One Case. Acta Paediatr. Taiwanica 2003, 44, 242–245. [Google Scholar]
- Hall, C.R.; Wu, Y.; Shaffer, L.G.; Hecht, J.T. Familial Case of Potocki - Shaffer Syndrome Associated with Microdeletion of EXT2 and ALX4. Clin. Genet. 2001, 60, 356–359. [Google Scholar] [CrossRef]
- Wuyts, W.; Di Gennaro, G.; Bianco, F.; Wauters, J.; Morocutti, C.; Pierelli, F.; Bossuyt, P.; Van Hul, W.; Casali, C. Molecular and Clinical Examination of an Italian DEFECT 11 Family. Eur. J. Hum. Genet. 1999, 7, 579–584. [Google Scholar] [CrossRef]
- Bartsch, O.; Wuyts, W.; Van Hul, W.; Hecht, J.T.; Meinecke, P.; Hogue, D.; Werner, W.; Zabel, B.; Hinkel, G.K.; Powell, C.M.; et al. Delineation of a Contiguous Gene Syndrome With Multiple Exostoses, Enlarged Parietal Foramina, Craniofacial Dysostosis, and Mental Retardation, Caused by Deletions in the Short Arm of Chromosome 11. Am. J. Hum. Genet. 1996, 58, 734–742. [Google Scholar]
- McCool, C.; Spinks-Franklin, A.; Noroski, L.M.; Potocki, L. Potocki–Shaffer Syndrome in a Child without Intellectual Disability—The Role of PHF21A in Cognitive Function. Am. J. Med. Genet. Part A 2017, 173, 716–720. [Google Scholar] [CrossRef]
- Labonne, J.D.J.; Vogt, J.; Reali, L.; Kong, I.K.; Layman, L.C.; Kim, H.G. A Microdeletion Encompassing PHF21A in an Individual with Global Developmental Delay and Craniofacial Anomalies. Am. J. Med. Genet. Part A 2015, 167, 3011–3018. [Google Scholar] [CrossRef]
- Sohn, Y.B.; Yim, S.Y.; Cho, E.H.; Kim, O.H. The First Korean Patient with Potocki-Shaffer Syndrome: A Rare Cause of Multiple Exostoses. J. Korean Med. Sci. 2015, 30, 214–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, N.D.; Turcott, C.M.; Tepperberg, J.H.; Mcdonald, M.T.; Aylsworth, A.S. A 137-Kb Deletion within the Potocki-Shaffer Syndrome Interval on Chromosome 11p11.2 Associated with Developmental Delay and Hypotonia. Am. J. Med. Genet. Part A 2013, 161, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Palka, C.; Alfonsi, M.; Mohn, A.; Guanciali Franchi, P.; Chiarelli, F.; Calabrese, G. Delayed Diagnosis of Potocki-Shaffer Syndrome in a Woman with Multiple Exostoses and Mental Retardation. Mol. Syndromol. 2012, 2, 259–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrarini, A.; Gaillard, M.; Guerry, F.; Ramelli, G.; Heidi, F.; Keddache, C.V.; Wieland, I.; Beckmann, J.S.; Jaquemont, S.; Martinet, D. Potocki-Shaffer Deletion Encompassing ALX4 in a Patient with Frontonasal Dysplasia Phenotype. Am. J. Med. Genet. 2014, 164, 346–352. [Google Scholar] [CrossRef]
- Yamamoto, T.; Akaboshi, S.; Ninomiya, H.; Nanba, E. DEFECT 11 Syndrome Associated with Agenesis of the Corpus Callosum. J. Med. Genet. 2001, 38, e5. [Google Scholar] [CrossRef] [Green Version]
- Francke, U.; George, D.L.; Brown, M.G.; Riccardi, V.M. Gene Dose Effect: Intraband Mapping of the LDH A Locus Using Cells from Four Individuals with Different Interstitial Deletions of 11p. Cytogenet. Cell Genet. 1977, 19, 197–207. [Google Scholar] [CrossRef]
- Abdul Jalil, M.F.; Russell, J.; Delatycki, M.; Gonzalvo, A. Congenital Biparietal Foramina Presenting with Multiple Concussions. Clin. Neurol. Neurosurg. 2016, 145, 6–7. [Google Scholar] [CrossRef]
- Wu, Y.Q.; Badano, J.L.; McCaskill, C.; Vogel, H.; Potocki, L.; Shaffer, L.G. Haploinsufficiency of ALX4 as a Potential Cause of Parietal Foramina in the 11p11.2 Contiguous Gene-Deletion Syndrome. Am. J. Hum. Genet. 2000, 67, 1327–1332. [Google Scholar] [CrossRef] [Green Version]
- Freeze, H.H. Human Disorders in N-Glycosylation and Animal Models. Biochim. Biophys. Acta 2002, 1573, 388–393. [Google Scholar] [CrossRef]
- Miller, B.S.; Freeze, H.H. New Disorders in Carbohydrate Metabolism: Congenital Disorders of Glycosylation and Their Impact on the Endocrine System. Rev. Endocr. Metab. Disord. 2003, 4, 103–113. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanaka, K.; Sugawara, Y.; Shimoji, T.; Nordtveit, T.I.; Kato, M.; Nakashima, M.; Saitsu, H.; Suzuki, T.; Yamakawa, K.; Aukrust, I.; et al. De Novo Truncating Variants in PHF21A Cause Intellectual Disability and Craniofacial Anomalies. Eur. J. Hum. Genet. 2019, 27, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.G.; Rosenfeld, J.A.; Scott, D.A.; Bénédicte, G.; Labonne, J.D.; Brown, J.; McGuire, M.; Mahida, S.; Naidu, S.; Gutierrez, J.; et al. Disruption of PHF21A Causes Syndromic Intellectual Disability with Craniofacial Anomalies, Epilepsy, Hypotonia, and Neurobehavioral Problems Including Autism. Mol. Autism 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- D’Alonzo, R.; Rigante, D.; Mencaroni, E.; Esposito, S. West Syndrome: A Review and Guide for Paediatricians. Clin. Drug Investig. 2018, 38, 113–124. [Google Scholar] [CrossRef]
- Jia, J.L.; Chen, S.; Sivarajah, V.; Stephens, D.; Cortez, M.A. Latitudinal Differences on the Global Epidemiology of Infantile Spasms: Systematic Review and Meta-Analysis. Orphanet J. Rare Dis. 2018, 13, 216. [Google Scholar] [CrossRef] [Green Version]
- Shields, W.D. Infantile Spasms: Little Seizures, BIG Consequences. Epilepsy Curr. 2006, 6, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.J.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 Histone Demethylase Activity by Its Associated Factors. Mol. Cell 2005, 19, 857–864. [Google Scholar] [CrossRef]
- Garay, P.M.; Wallner, M.A.; Iwase, S. Yin-Yang Actions of Histone Methylation Regulatory Complexes in the Brain. Epigenomics 2016, 8, 1689–1708. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. The Emerging Field of Epigenetics in Neurodegeneration and Neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef]
- Gerrard, D.T.; Berry, A.A.; Jennings, R.E.; Birket, M.J.; Zarrineh, P.; Garstang, M.G.; Withey, S.L.; Short, P.; Jiménez-Gancedo, S.; Firbas, P.N.; et al. Dynamic Changes in the Epigenomic Landscape Regulate Human Organogenesis and Link to Developmental Disorders. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and Its Corepressors Mediate Plasticity of Neuronal Gene Chromatin throughout Neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Collins, R.E.; De Cegli, R.; Alpatov, R.; Horton, J.R.; Shi, X.; Gozani, O.; Cheng, X.; Shi, Y. Recognition of Unmethylated Histone H3 Lysine 4 Links BHC80 to LSD1-Mediated Gene Repression. Nature 2007, 448, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Klajn, A.; Ferrai, C.; Stucchi, L.; Prada, I.; Podini, P.; Baba, T.; Rocchi, M.; Meldolesi, J.; D’Alessandro, R. The Rest Repression of the Neurosecretory Phenotype Is Negatively Modulated by BHC80, a Protein of the BRAF/HDAC Complex. J. Neurosci. 2009, 29, 6296–6307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakimi, M.A.; Bochar, D.A.; Chenoweth, J.; Lane, W.S.; Mandel, G.; Shiekhattar, R. A Core-BRAF35 Complex Containing Histone Deacetylase Mediates Repression of Neuronal-Specific Genes. Proc. Natl. Acad. Sci. USA 2002, 99, 7420–7425. [Google Scholar] [CrossRef] [Green Version]
- Porter, R.S.; Murata-Nakamura, Y.; Nagasu, H.; Kim, H.G.; Iwase, S. Transcriptome Analysis Revealed Impaired CAMP Responsiveness in PHF21A-Deficient Human Cells. Neuroscience 2018, 370, 170–180. [Google Scholar] [CrossRef]
- Takata, A.; Nakashima, M.; Saitsu, H.; Mizuguchi, T.; Mitsuhashi, S.; Takahashi, Y.; Okamoto, N.; Osaka, H.; Nakamura, K.; Tohyama, J.; et al. Comprehensive Analysis of Coding Variants Highlights Genetic Complexity in Developmental and Epileptic Encephalopathy. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Tatton-Brown, K.; Loveday, C.; Yost, S.; Clarke, M.; Ramsay, E.; Zachariou, A.; Elliott, A.; Wylie, H.; Ardissone, A.; Rittinger, O.; et al. Mutations in Epigenetic Regulation Genes Are a Major Cause of Overgrowth with Intellectual Disability. Am. J. Hum. Genet. 2017, 100, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Rockowitz, S.; Zheng, D. Significant Expansion of the REST/NRSF Cistrome in Human versus Mouse Embryonic Stem Cells: Potential Implications for Neural Development. Nucleic Acids Res. 2015, 43, 5730–5743. [Google Scholar] [CrossRef] [Green Version]
- Rockowitz, S.; Lien, W.H.; Pedrosa, E.; Wei, G.; Lin, M.; Zhao, K.; Lachman, H.M.; Fuchs, E.; Zheng, D. Comparison of REST Cistromes across Human Cell Types Reveals Common and Context-Specific Functions. PLoS Comput. Biol. 2014, 10, e1003671. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Lay, J.; Cheng, E.; Weng, J.; Sankar, R.; Baca, C.B. Recognition of Infantile Spasms Is Often Delayed: The ASSIST Study. J. Pediatr. 2017, 190, 215–221.e1. [Google Scholar] [CrossRef] [PubMed]
- Pavone, P.; Polizzi, A.; Marino, S.D.; Corsello, G.; Falsaperla, R.; Marino, S.; Ruggieri, M. West Syndrome: A Comprehensive Review. Neurol. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Decipher 286390 | Decipher 415213 | McCool | Labonne | Sohn | Kim GC14361 | Montgomery | Palka | Romeike | Bremond | Chuang Patient 1 | Chuang Patient 2 | Chuang Patient 3 | Wakui PSS03 | Wakui PSS04 | Wakui PSS08 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PSS | + | + | + | + | + | + & | + & | + & | + | + | ||||||||||||
| ALX/EXT4 | + | + | + | |||||||||||||||||||
| PHF21A | + | + | + | |||||||||||||||||||
| Birth parameters | ||||||||||||||||||||||
| SGA/Undergrowth | - | - | - | - | + | n.a | - | - | n.a | n.a | n.a | n.a | n.a | n.a | n.a | n.a | ||||||
| Appropriate | + | + | + | - | - | n.a | - | + | n.a | n.a | n.a | n.a | n.a | n.a | n.a | n.a | ||||||
| LGA/Overgrowth | - | - | - | + | - | n.a | + | - | n.a | n.a | n.a | n.a | n.a | n.a | n.a | n.a | ||||||
| Postnatal growth | ||||||||||||||||||||||
| Undergrowth | + | - | - | - | + | n.a | - | - | n.a | - † | n.a | n.a | n.a | + | + | n.a | ||||||
| Appropriate | - | - | - | - | - | n.a | - | + | n.a | + | n.a | n.a | n.a | - | - | n.a | ||||||
| Overgrowth | - | + | + | + | - | n.a | + | - | n.a | - | n.a | n.a | n.a | - | - | n.a | ||||||
| Neurodevelopment | ||||||||||||||||||||||
| Developmental delay | + | + | + » | + | + | + | + | + | + | - | + | - | - | - | + | + | ||||||
| Intellectual disability | + | + | - | - | + | - | + | + | + | + | - | + | + | - | - | - | ||||||
| Language delay | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||||||
| Neurological findings | ||||||||||||||||||||||
| Hypotonia | + | - | - | - | + | + | + | - | - | - | - | - | - | - | + | - | ||||||
| Epilepsy | + | + | - | - | - | - ° | - | + | + * | - | - | + | + | - | - | - | ||||||
| MRI findings | ||||||||||||||||||||||
| Corpus callosum | + | - | - | - | - | - | - | - | + | - | - | - | - | - | - | + | ||||||
| Prominent CSF spaces | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | ||||||
| Other brain anomalies | - | + | - | - | - | - | - | - | + | - | - | - | - | - | - | - | ||||||
| Genitourinary | ||||||||||||||||||||||
| Micropenis | - | - | - | + | - | - | - | - | - | - | + | - | - | - | + | - | ||||||
| Cryptorchidism | - | - | - | - | - | + | - | - | + | - | - | - | + | - | + | - | ||||||
| Ocular anomalies | ||||||||||||||||||||||
| Cataract | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | ||||||
| Strabismus | - | - | + | - | + | - | - | - | - | - | + | - | - | - | - | - | ||||||
| Nystagmus | - | - | - | - | + | - | - | - | - | + | - | - | - | - | - | - | ||||||
| Hearing anomalies | ||||||||||||||||||||||
| hearing loss | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | ||||||
| Other | IgA deficiency; sleep apnea | Café-au-lait spots | Recurrent infections | Ptosis | Pectus excavatum. Recurrent otitis media | Pectus excavatum. | Cardiomyopathy; Osteochondromas; Anemia | Bilateral aniridia; Kidney tumor; Obesity | Umbilical hernia | Bowing of lower extremities | Umbilical hernia; Myopia; Recurrent infections | Wilms’ tumor; Aniridia | ||||||||||
| Wakui PSS10 | Wakui PSS12 | Wakui PSS13 | Wuyts patient 1 | Wuyts patient 2 | Wuyts patient 3 | Chien 3 patients | Hall 3 patients | Wuyts 4 patients | Bartsch Patient 2 | Bartsch Patient 6–8 | Potocky | McGaughran | Shaffer III-1 | Shaffer III-2 | Shaffer II-4 | |||||||
| PSS | + | + | + | + | + | + | + | + | + | + | + | + | ||||||||||
| ALX/EXT4 | + | + | + | + | ||||||||||||||||||
| PHF21A | ||||||||||||||||||||||
| Birth parameters | ||||||||||||||||||||||
| SGA/Undergrowth | n.a | - | + | - | - | - | n.a | n.a | n.a | - | - | - | - | + ^ | n.a | - | ||||||
| Appropriate | n.a | + | - | + | + | + | n.a | n.a | n.a | + | + | + | + | - | n.a | + ^ | ||||||
| LGA/Overgrowth | n.a | - | - | - | - | - | n.a | n.a | n.a | - | - | - | - | - | n.a | - | ||||||
| Postnatal growth | ||||||||||||||||||||||
| Undergrowth | - | - | + | - | - | + | n.a | n.a | n.a | - | - | - | - | + ^ | n.a | - | ||||||
| Appropriate | + | + | - | + | + | - | n.a | n.a | n.a | + | + | + | + | - | n.a | + ^ | ||||||
| Overgrowth | - | - | - | - | - | - | n.a | n.a | n.a | - | - | - | - | - | n.a | - | ||||||
| Neurodevelopment | ||||||||||||||||||||||
| Developmental delay | - | + | + | + | + | + | + | - | - | - | - | + | - | + ^ | - | - | ||||||
| Intellectual disability | - | - | - | + | + | + | + | - | - | + | - | + | + | + | + | + | ||||||
| Language delay | + | - | - | + | - | - | - | - | - | - | - | - | - | + ^ | - | - | ||||||
| Neurological findings | ||||||||||||||||||||||
| Hypotonia | - | - | + | + | + | + | + | - | - | + | - | - | - | + | + | + | ||||||
| Epilepsy | - | - | - | + | - | + | - º | - | + ΅ | + | - | - | - | + ^ | - | + | ||||||
| MRI findings | ||||||||||||||||||||||
| Corpus callosum | + | + | - | - | - | - | + | - | - | - | - | - | - | - | - | - | ||||||
| Prominent CSF spaces | - | - | + | - | - | - | + | - | - | - | - | - | - | + ^ | - | - | ||||||
| Other brain anomalies | - | + | - | + | - | + | - | - | + ΅ | - | - | - | - | + ^ | - | + | ||||||
| Genitourinary | ||||||||||||||||||||||
| Micropenis | + | - | - | - | - | + | + º | - | - | + | - | - | + | - | - | + | ||||||
| Cryptorchidism | - | - | + | - | - | - | - º | - | - | - | - | - | + | - | - | + | ||||||
| Ocular anomalies | ||||||||||||||||||||||
| Cataract | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | ||||||
| Strabismus | + | - | + | - | + | + | + | - | - | + | - | + | - | + | - | + | ||||||
| Nystagmus | - | - | - | - | + | + | - | - | - | + | - | - | - | - | - | - | ||||||
| Hearing anomalies | ||||||||||||||||||||||
| hearing loss | - | + | + | - | - | - | - | - | - | - | - | - | - | - | + ^ | |||||||
| Other | Deceased from multiorgan failure | Small testis | VSD; Recurrent infections | High myopia; Obesity | Anal atresia and fistula; VSD | Myopia; Acrocephalosyndactyly; Adipose | 7625 Asthma; Hyperactivity | Adipose appearance | Café-au-lait spots Capillary hemangioma | Bilateral ptosis | Aniridia; Wilms’ tumor; Short stature | Borderline hypothyroidism; Simian crease | borderline high TSH; Simian crease; Obese | Adipose appearance; Aggressive behavior | ||||||||
| Dysmorphology | n | % | PSS | ALX/EXT4 | PHF21A |
|---|---|---|---|---|---|
| Brachycephaly | 17 | 61 | 14 | 2 | 1 |
| Broad forehead | 12 | 43 | 9 | 1 | 1 |
| Epicanthus | 12 | 43 | 9 | 1 | 2 |
| Downturned mouth | 11 | 39 | 11 | 0 | 0 |
| High forehead | 10 | 36 | 8 | 2 | 0 |
| Prominent nasal bridge | 10 | 36 | 10 | 0 | 0 |
| Sparse lateral eyebrows | 9 | 32 | 8 | 1 | 0 |
| Short philtrum | 9 | 32 | 7 | 1 | 1 |
| Microcephaly | 8 | 29 | 8 | 0 | 0 |
| Hypoplastic nares | 6 | 21 | 5 | 1 | 0 |
| Broad nasal tip | 5 | 18 | 5 | 0 | 0 |
| Low set ears | 5 | 18 | 4 | 1 | 0 |
| Large/protuberant ears | 5 | 18 | 3 | 1 | 1 |
| Telecanthus | 4 | 14 | 4 | 0 | 0 |
| Upslanting palpebral fissures | 4 | 14 | 4 | 0 | 0 |
| Thin lips | 4 | 14 | 3 | 0 | 1 |
| Micrognathia | 4 | 14 | 2 | 1 | 1 |
| Turricephaly | 3 | 11 | 2 | 0 | 1 |
| Broad nasal bridge | 3 | 11 | 2 | 0 | 1 |
| Short neck | 3 | 11 | 3 | 0 | 0 |
| Small nose | 2 | 7 | 2 | 0 | 0 |
| Small mouth | 2 | 7 | 1 | 0 | 1 |
| Full cheeks | 2 | 7 | 0 | 1 | 1 |
| Prominent chin | 2 | 7 | 1 | 1 | 0 |
| Downslanted palpebral fissures | 1 | 3 | 0 | 1 | 0 |
| TOTAL | 28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trajkova, S.; Di Gregorio, E.; Ferrero, G.B.; Carli, D.; Pavinato, L.; Delplancq, G.; Kuentz, P.; Brusco, A. New Insights into Potocki-Shaffer Syndrome: Report of Two Novel Cases and Literature Review. Brain Sci. 2020, 10, 788. https://doi.org/10.3390/brainsci10110788
Trajkova S, Di Gregorio E, Ferrero GB, Carli D, Pavinato L, Delplancq G, Kuentz P, Brusco A. New Insights into Potocki-Shaffer Syndrome: Report of Two Novel Cases and Literature Review. Brain Sciences. 2020; 10(11):788. https://doi.org/10.3390/brainsci10110788
Chicago/Turabian StyleTrajkova, Slavica, Eleonora Di Gregorio, Giovanni Battista Ferrero, Diana Carli, Lisa Pavinato, Geoffroy Delplancq, Paul Kuentz, and Alfredo Brusco. 2020. "New Insights into Potocki-Shaffer Syndrome: Report of Two Novel Cases and Literature Review" Brain Sciences 10, no. 11: 788. https://doi.org/10.3390/brainsci10110788
APA StyleTrajkova, S., Di Gregorio, E., Ferrero, G. B., Carli, D., Pavinato, L., Delplancq, G., Kuentz, P., & Brusco, A. (2020). New Insights into Potocki-Shaffer Syndrome: Report of Two Novel Cases and Literature Review. Brain Sciences, 10(11), 788. https://doi.org/10.3390/brainsci10110788