
β-Hydroxybutyrate, a Ketone Body, Potentiates the Antioxidant Defense via Thioredoxin 1 Upregulation in Cardiomyocytes


Abstract
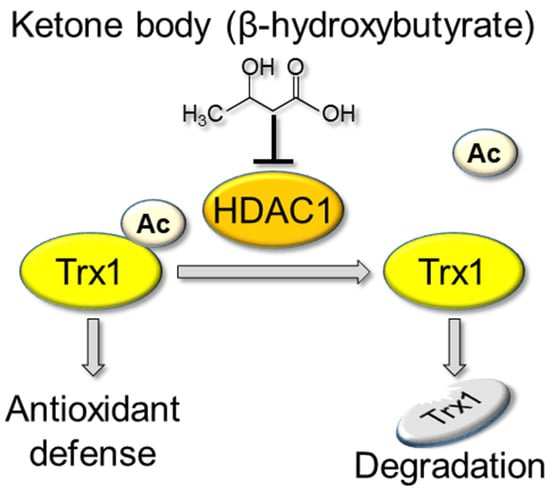
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Primary Cultures of Neonatal Cardiomyocytes and Fibroblasts
2.2. Immunoblot Analyses
2.3. Adenovirus Vectors
2.4. Ketogenic Diet
2.5. Statistical Methods
3. Results
3.1. βHB Upregulates Trx1
3.2. βHB Confers Resistance against Oxidative Stress through a Trx1-Dependent Manner
3.3. βHB Prevents H2O2-Induced mTOR Inhibition in a Trx1-Dependent Manner
3.4. βHB Prevents H2O2-Induced AMPK Inhibition in a Trx1-Dependent Manner
3.5. βHB May Upregulate Trx1 through HDAC Inhibition
4. Discussion
4.1. βHB Upregulates Trx1
4.2. Ketone Bodies in mTOR Regulation
4.3. Ketone Body in Diabetic Cardiomyopathy
4.4. Ketone Bodies, HDAC Inhibtioin and Trx1 as a Longevity Factor
4.5. Mechanism Responsible for Trx1 Upregulation
4.6. Experimental Limitation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engin, A. The Definition and Prevalence of Obesity and Metabolic Syndrome. Adv. Exp. Med. Biol. 2017, 960, 1–17. [Google Scholar] [CrossRef]
- Paolillo, S.; Marsico, F.; Prastaro, M.; Renga, F.; Esposito, L.; De Martino, F.; Di Napoli, P.; Esposito, I.; Ambrosio, A.; Ianniruberto, M.; et al. Diabetic Cardiomyopathy: Definition, Diagnosis, and Therapeutic Implications. Heart Fail. Clin. 2019, 15, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, S.; Hsu, C.P.; Sadoshima, J. Regulation of cell survival and death by pyridine nucleotides. Circ. Res. 2012, 111, 611–627. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, S.I.; Hirata, T.; Suzuki, W.; Naito, D.; Chen, Y.; Chin, A.; Yaginuma, H.; Saito, T.; Nagarajan, N.; Zhai, P.; et al. Thioredoxin-1 maintains mechanistic target of rapamycin (mTOR) function during oxidative stress in cardiomyocytes. J. Biol. Chem. 2017, 292, 18988–19000. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Oka, S.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.I.; Byun, J.; Huang, C.Y.; Imai, N.; Ralda, G.E.; Zhai, P.; Xu, X.; Kashyap, S.S.; Warren, J.S.; Maschek, J.A.; et al. Nampt Potentiates Antioxidant Defense in Diabetic Cardiomyopathy. Circ. Res. 2021. [Google Scholar] [CrossRef]
- Abdul Kadir, A.; Clarke, K.; Evans, R.D. Cardiac ketone body metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165739. [Google Scholar] [CrossRef]
- Nakamura, M.; Odanovic, N.; Nakada, Y.; Dohi, S.; Zhai, P.; Ivessa, A.; Yang, Z.; Abdellatif, M.; Sadoshima, J. Dietary Carbohydrates Restriction Inhibits the Development of Cardiac Hypertrophy and Heart Failure. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Yurista, S.R.; Matsuura, T.R.; Sillje, H.H.W.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ. Heart Fail. 2021, 14, e007684. [Google Scholar] [CrossRef]
- Nikolic, M.; Zivkovic, V.; Jovic, J.J.; Sretenovic, J.; Davidovic, G.; Simovic, S.; Djokovic, D.; Muric, N.; Bolevich, S.; Jakovljevic, V. SGLT2 inhibitors: A focus on cardiac benefits and potential mechanisms. Heart Fail. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.L.; Zhang, L.; Wagg, C.; Al Batran, R.; Gopal, K.; Levasseur, J.; Leone, T.; Dyck, J.R.B.; Ussher, J.R.; Muoio, D.M.; et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 2019, 115, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, M.; Toh, R.; Irino, Y.; Mori, T.; Nakajima, H.; Hara, T.; Honjo, T.; Satomi-Kobayashi, S.; Shinke, T.; Tanaka, H.; et al. beta-Hydroxybutyrate elevation as a compensatory response against oxidative stress in cardiomyocytes. Biochem. Biophys. Res. Commun. 2016, 475, 322–328. [Google Scholar] [CrossRef]
- Uchihashi, M.; Hoshino, A.; Okawa, Y.; Ariyoshi, M.; Kaimoto, S.; Tateishi, S.; Ono, K.; Yamanaka, R.; Hato, D.; Fushimura, Y.; et al. Cardiac-Specific Bdh1 Overexpression Ameliorates Oxidative Stress and Cardiac Remodeling in Pressure Overload-Induced Heart Failure. Circ. Heart Fail. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, Y.Q.; Wu, C.H.; Zhang, Y.L.; Zhao, S.T.; Chen, Y.J.; Deng, Y.H.; Xuan, A.; Sun, X.D. The effect of ketogenic diet on behaviors and synaptic functions of naive mice. Brain Behav. 2019, 9, e01246. [Google Scholar] [CrossRef]
- Guerci, B.; Benichou, M.; Floriot, M.; Bohme, P.; Fougnot, S.; Franck, P.; Drouin, P. Accuracy of an electrochemical sensor for measuring capillary blood ketones by fingerstick samples during metabolic deterioration after continuous subcutaneous insulin infusion interruption in type 1 diabetic patients. Diabetes Care 2003, 26, 1137–1141. [Google Scholar] [CrossRef] [Green Version]
- Galic, S.; Loh, K.; Murray-Segal, L.; Steinberg, G.R.; Andrews, Z.B.; Kemp, B.E. AMPK signaling to acetyl-CoA carboxylase is required for fasting- and cold-induced appetite but not thermogenesis. Elife 2018, 7. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, F.; Wang, J.; Zhang, D.; Zhao, X. Ketogenic diet attenuates aging-associated myocardial remodeling and dysfunction in mice. Exp. Gerontol. 2020, 140, 111058. [Google Scholar] [CrossRef]
- Rolfe, M.; McLeod, L.E.; Pratt, P.F.; Proud, C.G. Activation of protein synthesis in cardiomyocytes by the hypertrophic agent phenylephrine requires the activation of ERK and involves phosphorylation of tuberous sclerosis complex 2 (TSC2). Biochem. J. 2005, 388, 973–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Honda, M.; Takabatake, T. Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J. Am. Coll. Cardiol. 2001, 37, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Brooke, J.; Stiell, M.; Ojo, O. Evaluation of the Accuracy of Capillary Hydroxybutyrate Measurement Compared with Other Measurements in the Diagnosis of Diabetic Ketoacidosis: A Systematic Review. Int. J. Environ. Res. Public Health 2016, 13, 837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.K.; Kim, D.H.; Bang, E.; Choi, Y.J.; Chung, H.Y. beta-Hydroxybutyrate Suppresses Lipid Accumulation in Aged Liver through GPR109A-mediated Signaling. Aging Dis. 2020, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Kopp, Z.A.; Hsieh, J.L.; Li, A.; Wang, W.; Bhatt, D.T.; Lee, A.; Kim, S.Y.; Fan, D.; Shah, V.; Siddiqui, E.; et al. Heart-specific Rpd3 downregulation enhances cardiac function and longevity. Aging 2015, 7, 648–663. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab. 2017, 26, 547–557.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, V.I.; Cortez, L.A.; Lew, C.M.; Rodriguez, M.; Webb, C.R.; Van Remmen, H.; Chaudhuri, A.; Qi, W.; Lee, S.; Bokov, A.; et al. Thioredoxin 1 overexpression extends mainly the earlier part of life span in mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66, 1286–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Zouhir, S.; Bernal-Bayard, J.; Cordero-Alba, M.; Cardenal-Munoz, E.; Guimaraes, B.; Lazar, N.; Ramos-Morales, F.; Nessler, S. The structure of the Slrp-Trx1 complex sheds light on the autoinhibition mechanism of the type III secretion system effectors of the NEL family. Biochem. J. 2014, 464, 135–144. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oka, S.-i.; Tang, F.; Chin, A.; Ralda, G.; Xu, X.; Hu, C.; Yang, Z.; Abdellatif, M.; Sadoshima, J. β-Hydroxybutyrate, a Ketone Body, Potentiates the Antioxidant Defense via Thioredoxin 1 Upregulation in Cardiomyocytes. Antioxidants 2021, 10, 1153. https://doi.org/10.3390/antiox10071153
Oka S-i, Tang F, Chin A, Ralda G, Xu X, Hu C, Yang Z, Abdellatif M, Sadoshima J. β-Hydroxybutyrate, a Ketone Body, Potentiates the Antioxidant Defense via Thioredoxin 1 Upregulation in Cardiomyocytes. Antioxidants. 2021; 10(7):1153. https://doi.org/10.3390/antiox10071153
Chicago/Turabian StyleOka, Shin-ichi, Fan Tang, Adave Chin, Guersom Ralda, Xiaoyong Xu, Chengchen Hu, Zhi Yang, Maha Abdellatif, and Junichi Sadoshima. 2021. "β-Hydroxybutyrate, a Ketone Body, Potentiates the Antioxidant Defense via Thioredoxin 1 Upregulation in Cardiomyocytes" Antioxidants 10, no. 7: 1153. https://doi.org/10.3390/antiox10071153
APA StyleOka, S. -i., Tang, F., Chin, A., Ralda, G., Xu, X., Hu, C., Yang, Z., Abdellatif, M., & Sadoshima, J. (2021). β-Hydroxybutyrate, a Ketone Body, Potentiates the Antioxidant Defense via Thioredoxin 1 Upregulation in Cardiomyocytes. Antioxidants, 10(7), 1153. https://doi.org/10.3390/antiox10071153