

The Hepatic Mitochondrial Alterations Exacerbate Meta-Inflammation in Autism Spectrum Disorders

 ,
, 

 , ,
, ,  ,
,  , ,
, ,  ,
,  ,
,  and
and 
Abstract
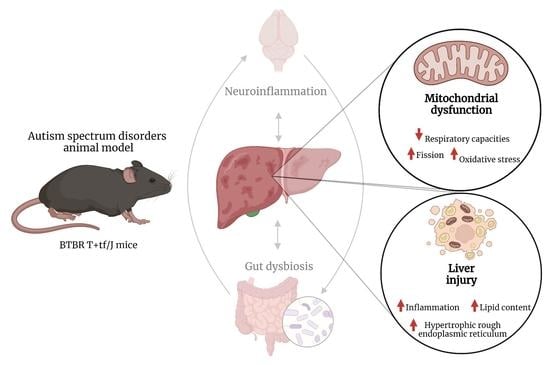
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animal and Experimental Design
2.3. Body Composition and Energy Balance
2.4. Evaluation of Metabolic Parameters and Inflammatory Markers in Serum and Tissue
2.5. Hepatic Histological Analyses
2.6. Western Blot Analysis
2.7. Mitochondrial Preparation and Analysis
2.8. Hepatic Oxidative Stress, Antioxidant/Detoxifying Defenses and Lipid Peroxidation
2.9. Statistical Analysis
3. Results
3.1. Metabolic and Inflammatory Profile in BTBR Mice
3.2. Hepatic Morphological Features in BTBR Mice
3.3. Hepatic Markers of ER Stress and Mitochondrial Dynamics in BTBR Mice
3.4. Hepatic Mitochondrial Oxidative Capacities in BTBR Mice
3.5. Hepatic Oxidative Stress and Antioxidant/Detoxifying Defense in BTBR Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association (APA). Diagnostic and Statistical Manual of Mental Disorders: DSM-5, DSM-5, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013; ISBN 9780890425541. [Google Scholar]
- Abrahams, B.S.; Geschwind, D.H. Connecting Genes to Brain in the Autism Spectrum Disorders. Arch. Neurol. 2010, 67, 395–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristiano, C.; Lama, A.; Lembo, F.; Mollica, M.P.; Calignano, A.; Mattace Raso, G. Interplay Between Peripheral and Central Inflammation in Autism Spectrum Disorders: Possible Nutritional and Therapeutic Strategies. Front. Physiol. 2018, 9, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwood, P.; Wills, S.; de Water, J. The Immune Response in Autism: A New Frontier for Autism Research. J. Leukoc. Biol. 2006, 80, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial Activation and Increased Microglial Density Observed in the Dorsolateral Prefrontal Cortex in Autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; de Water, J. Elevated Plasma Cytokines in Autism Spectrum Disorders Provide Evidence of Immune Dysfunction and Are Associated with Impaired Behavioral Outcome. Brain Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Ricci, S.; Businaro, R.; Ippoliti, F.; lo Vasco, V.R.; Massoni, F.; Onofri, E.; Troili, G.M.; Pontecorvi, V.; Morelli, M.; Rapp Ricciardi, M.; et al. Altered Cytokine and BDNF Levels in Autism Spectrum Disorder. Neurotox. Res. 2013, 24, 491–501. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial Activation and Neuroinflammation in the Brain of Patients with Autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Hernández-Aguilera, A.; Rull, A.; Rodríguez-Gallego, E.; Riera-Borrull, M.; Luciano-Mateo, F.; Camps, J.; Menéndez, J.A.; Joven, J. Mitochondrial Dysfunction: A Basic Mechanism in Inflammation-Related Non-Communicable Diseases and Therapeutic Opportunities. Mediators Inflamm. 2013, 2013, 135698. [Google Scholar] [CrossRef] [Green Version]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell. Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef]
- Buro, A.W.; Salinas-Miranda, A.; Marshall, J.; Gray, H.L.; Kirby, R.S. Autism Spectrum Disorder Diagnosis and Other Child, Family, and Community Risk Factors for Obesity among Children and Adolescents Aged Ten to Seventeen Years in the United States: A Mediation Analysis. Child. Obes. 2022. [Google Scholar] [CrossRef]
- Russo, R.; Cristiano, C.; Avagliano, C.; de Caro, C.; la Rana, G.; Raso, G.M.; Canani, R.B.; Meli, R.; Calignano, A. Gut-Brain Axis: Role of Lipids in the Regulation of Inflammation, Pain and CNS Diseases. Curr. Med. Chem. 2018, 25, 3930–3952. [Google Scholar] [CrossRef] [PubMed]
- Coretti, L.; Cristiano, C.; Florio, E.; Scala, G.; Lama, A.; Keller, S.; Cuomo, M.; Russo, R.; Pero, R.; Paciello, O.; et al. Sex-Related Alterations of Gut Microbiota Composition in the BTBR Mouse Model of Autism Spectrum Disorder. Sci. Rep. 2017, 7, 45356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, M.S.; Newell, C.; Bomhof, M.R.; Reimer, R.A.; Hittel, D.S.; Rho, J.M.; Vogel, H.J.; Shearer, J. Metabolomic Modeling To Monitor Host Responsiveness to Gut Microbiota Manipulation in the BTBR(T+tf/j) Mouse. J. Proteome Res. 2016, 15, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.; Lama, A.; Pirozzi, C.; Cavaliere, G.; Trinchese, G.; di Guida, F.; Nitrato Izzo, A.; Cimmino, F.; Paciello, O.; de Biase, D.; et al. Palmitoylethanolamide Counteracts Hepatic Metabolic Inflexibility Modulating Mitochondrial Function and Efficiency in Diet-Induced Obese Mice. FASEB J. 2020, 34, 350–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver Immunology and Its Role in Inflammation and Homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrad, D.; Wueest, S. The Gut-Adipose-Liver Axis in the Metabolic Syndrome. Physiology 2014, 29, 304–313. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorders: A Systematic Review and Meta-Analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef] [Green Version]
- Newell, C.; Shutt, T.E.; Ahn, Y.; Hittel, D.S.; Khan, A.; Rho, J.M.; Shearer, J. Tissue Specific Impacts of a Ketogenic Diet on Mitochondrial Dynamics in the BTBR. Front. Physiol. 2016, 7, 654. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorder: Unique Abnormalities and Targeted Treatments. Semin. Pediatr. Neurol. 2020, 35, 100829. [Google Scholar] [CrossRef]
- Cristiano, C.; Pirozzi, C.; Coretti, L.; Cavaliere, G.; Lama, A.; Russo, R.; Lembo, F.; Mollica, M.P.; Meli, R.; Calignano, A.; et al. Palmitoylethanolamide Counteracts Autistic-like Behaviours in BTBR T+tf/J Mice: Contribution of Central and Peripheral Mechanisms. Brain Behav. Immun. 2018, 74, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Chauhan, V.; Kaur, K.; Brown, W.T.; LaFauci, G.; Wegiel, J.; Chauhan, A. Alterations in Mitochondrial DNA Copy Number and the Activities of Electron Transport Chain Complexes and Pyruvate Dehydrogenase in the Frontal Cortex from Subjects with Autism. Transl. Psychiatry 2013, 3, e299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.; Wynne, R.; Frye, R.E.; Melnyk, S.; James, S.J. Increased Susceptibility to Ethylmercury-Induced Mitochondrial Dysfunction in a Subset of Autism Lymphoblastoid Cell Lines. J. Toxicol. 2015, 2015, 573701. [Google Scholar] [CrossRef]
- McFarlane, H.G.; Kusek, G.K.; Yang, M.; Phoenix, J.L.; Bolivar, V.J.; Crawley, J.N. Autism-like Behavioral Phenotypes in BTBR T+tf/J Mice. Genes Brain Behav. 2008, 7, 152–163. [Google Scholar] [CrossRef]
- Bartok, A.; Weaver, D.; Golenár, T.; Nichtova, Z.; Katona, M.; Bánsághi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 Receptor Isoforms Differently Regulate ER-Mitochondrial Contacts and Local Calcium Transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y.; Sabouny, R.; Villa, B.R.; Yee, N.C.; Mychasiuk, R.; Uddin, G.M.; Rho, J.M.; Shutt, T.E. Aberrant Mitochondrial Morphology and Function in the BTBR Mouse Model of Autism Is Improved by Two Weeks of Ketogenic Diet. Int. J. Mol. Sci. 2020, 21, 3266. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Dufour, J.-F.; Shibao, K.; Knickelbein, R.; O’Neill, A.F.; Bode, H.-P.; Cassio, D.; St-Pierre, M.V.; LaRusso, N.F.; Leite, M.F.; et al. Regulation of Ca2+ Signaling in Rat Bile Duct Epithelia by Inositol 1,4,5-Trisphosphate Receptor Isoforms. Hepatology 2002, 36, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, K.; Pusl, T.; O’Neill, A.F.; Dranoff, J.A.; Nathanson, M.H. The Type II Inositol 1,4,5-Trisphosphate Receptor Can Trigger Ca2+ Waves in Rat Hepatocytes. Gastroenterology 2002, 122, 1088–1100. [Google Scholar] [CrossRef]
- Khamphaya, T.; Chukijrungroat, N.; Saengsirisuwan, V.; Mitchell-Richards, K.A.; Robert, M.E.; Mennone, A.; Ananthanarayanan, M.; Nathanson, M.H.; Weerachayaphorn, J. Nonalcoholic Fatty Liver Disease Impairs Expression of the Type II Inositol 1,4,5-trisphosphate Receptor. Hepatology 2018, 67, 560–574. [Google Scholar] [CrossRef] [Green Version]
- Shibao, K.; Hirata, K.; Robert, M.E.; Nathanson, M.H. Loss of Inositol 1,4,5-Trisphosphate Receptors from Bile Duct Epithelia Is a Common Event in Cholestasis. Gastroenterology 2003, 125, 1175–1187. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid. Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinchese, G.; Cavaliere, G.; Cimmino, F.; Catapano, A.; Carta, G.; Pirozzi, C.; Murru, E.; Lama, A.; Meli, R.; Bergamo, P.; et al. Decreased Metabolic Flexibility in Skeletal Muscle of Rat Fed with a High-Fat Diet Is Recovered by Individual CLA Isomer Supplementation via Converging Protective Mechanisms. Cells 2020, 9, 823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, F.; Catapano, A.; Trinchese, G.; Cavaliere, G.; Culurciello, R.; Fogliano, C.; Penna, E.; Lucci, V.; Crispino, M.; Avallone, B.; et al. Dietary Micronutrient Management to Treat Mitochondrial Dysfunction in Diet-Induced Obese Mice. Int. J. Mol. Sci. 2021, 22, 2862. [Google Scholar] [CrossRef]
- Cairns, C.B.; Walther, J.; Harken, A.H.; Banerjee, A. Mitochondrial Oxidative Phosphorylation Thermodynamic Efficiencies Reflect Physiological Organ Roles. Am. J. Physiol. 1998, 274, R1376–R1383. [Google Scholar] [CrossRef]
- Barja, G. Mitochondrial Free Radical Production and Aging in Mammals and Birds. Ann. N. Y. Acad. Sci. 1998, 854, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Hausladen, A.; Fridovich, I. Measuring Nitric Oxide and Superoxide: Rate Constants for Aconitase Reactivity. Methods Enzym. 1996, 269, 37–41. [Google Scholar] [CrossRef]
- Flohé, L.; Otting, F. Superoxide Dismutase Assays. Methods Enzym. 1984, 105, 93–104. [Google Scholar] [CrossRef]
- Viggiano, E.; Mollica, M.P.; Lionetti, L.; Cavaliere, G.; Trinchese, G.; de Filippo, C.; Chieffi, S.; Gaita, M.; Barletta, A.; de Luca, B.; et al. Effects of an High-Fat Diet Enriched in Lard or in Fish Oil on the Hypothalamic Amp-Activated Protein Kinase and Inflammatory Mediators. Front. Cell. Neurosci. 2016, 10, 150. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Gevezova, M.; Sarafian, V.; Anderson, G.; Maes, M. Inflammation and Mitochondrial Dysfunction in Autism Spectrum Disorder. CNS Neurol. Disord. Drug Targets 2020, 19, 320–333. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Crispino, M.; Trinchese, G.; Penna, E.; Cimmino, F.; Catapano, A.; Villano, I.; Perrone-Capano, C.; Mollica, M.P. Interplay between Peripheral an.nd Central Inflammation in Obesity-Promoted Disorders: The Impact on Synaptic Mitochondrial Functions. Int. J. Mol. Sci. 2020, 21, 5964. [Google Scholar] [CrossRef] [PubMed]
- Valleau, J.C.; Sullivan, E.L. The Impact of Leptin on Perinatal Development and Psychopathology. J. Chem. Neuroanat. 2014, 61–62, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin Stimulates Glucose Utilization and Fatty-Acid Oxidation by Activating AMP-Activated Protein Kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.M.; Doss, H.M.; Kim, K.S. Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int. J. Mol. Sci. 2020, 21, 1219. [Google Scholar] [CrossRef] [Green Version]
- Ashwood, P.; Corbett, B.A.; Kantor, A.; Schulman, H.; de Water, J.; Amaral, D.G. In Search of Cellular Immunophenotypes in the Blood of Children with Autism. PLoS ONE 2011, 6, e19299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Roussin, L.; Prince, N.; Perez-Pardo, P.; Kraneveld, A.D.; Rabot, S.; Naudon, L. Role of the Gut Microbiota in the Pathophysiology of Autism Spectrum Disorder: Clinical and Preclinical Evidence. Microorganisms 2020, 8, 1369. [Google Scholar] [CrossRef]
- Kong, C.; Gao, R.; Yan, X.; Huang, L.; Qin, H. Probiotics Improve Gut Microbiota Dysbiosis in Obese Mice Fed a High-Fat or High-Sucrose Diet. Nutrition 2019, 60, 175–184. [Google Scholar] [CrossRef]
- Leduc, E.H.; Wilson, J.W. An Electron Microscope Study of Intranuclear Inclusions in Mouse Liver and Hepatoma. J. Biophys. Biochem. Cytol. 1959, 6, 427–430. [Google Scholar] [CrossRef]
- Schwertheim, S.; Kälsch, J.; Jastrow, H.; Schaefer, C.M.; Theurer, S.; Ting, S.; Canbay, A.; Wedemeyer, H.; Schmid, K.W.; Baba, H.A. Characterization of Two Types of Intranuclear Hepatocellular Inclusions in NAFLD. Sci. Rep. 2020, 10, 16533. [Google Scholar] [CrossRef] [PubMed]
- Aravinthan, A.; Verma, S.; Coleman, N.; Davies, S.; Allison, M.; Alexander, G. Vacuolation in Hepatocyte Nuclei Is a Marker of Senescence. J. Clin. Pathol. 2012, 65, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T.; Bièche, I.; Mansouri, A.; Laurendeau, I.; Cazals-Hatem, D.; Feldmann, G.; Bedossa, P.; Paradis, V.; Martinot-Peignoux, M.; Lebrec, D.; et al. In Vivo Hepatic Endoplasmic Reticulum Stress in Patients with Chronic Hepatitis C. J. Pathol. 2010, 221, 264–274. [Google Scholar] [CrossRef]
- Wang, P.T.; Garcin, P.O.; Fu, M.; Masoudi, M.; St-Pierre, P.; Panté, N.; Nabi, I.R. Distinct Mechanisms Controlling Rough and Smooth Endoplasmic Reticulum Contacts with Mitochondria. J. Cell Sci. 2015, 128, 2759–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, E.; Rieusset, J. Metabolic Signaling Functions of ER–Mitochondria Contact Sites: Role in Metabolic Diseases. J. Mol. Endocrinol. 2017, 58, R87–R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Patergnani, S.; Pinton, P. The Endoplasmic Reticulum-Mitochondria Connection: One Touch, Multiple Functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Galloway, C.A.; Lee, H.; Brookes, P.S.; Yoon, Y. Decreasing Mitochondrial Fission Alleviates Hepatic Steatosis in a Murine Model of Nonalcoholic Fatty Liver Disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G632–G641. [Google Scholar] [CrossRef] [Green Version]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the Endoplasmic Reticulum-Mitochondria Connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51. [Google Scholar] [CrossRef]
- Carrasco, M.; Salazar, C.; Tiznado, W.; Ruiz, L.M. Alterations of Mitochondrial Biology in the Oral Mucosa of Chilean Children with Autism Spectrum Disorder (ASD). Cells 2019, 8, 367. [Google Scholar] [CrossRef]
- Anitha, A.; Nakamura, K.; Thanseem, I.; Yamada, K.; Iwayama, Y.; Toyota, T.; Matsuzaki, H.; Miyachi, T.; Yamada, S.; Tsujii, M.; et al. Brain Region-Specific Altered Expression and Association of Mitochondria-Related Genes in Autism. Mol. Autism 2012, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Gutierrez Rios, P.; Kuo, S.-H.; Akman, H.O.; Rosoklija, G.; Tanji, K.; Dwork, A.; Schon, E.A.; DiMauro, S.; Goldman, J.; et al. Mitochondrial Abnormalities in Temporal Lobe of Autistic Brain. Neurobiol. Dis. 2013, 54, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.A.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; et al. Nutritional and Metabolic Status of Children with Autism vs. Neurotypical Children, and the Association with Autism Severity. Nutr. Metab. 2011, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Pecorelli, A.; Ferrara, F.; Messano, N.; Cordone, V.; Schiavone, M.L.; Cervellati, F.; Woodby, B.; Cervellati, C.; Hayek, J.; Valacchi, G. Alterations of Mitochondrial Bioenergetics, Dynamics, and Morphology Support the Theory of Oxidative Damage Involvement in Autism Spectrum Disorder. FASEB J. 2020, 34, 6521–6538. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Uncoupling: New Approaches to an Old Problem of Bioenergetics. Biochim. Biophys. Acta 1998, 1363, 100–124. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Zhang, D.M.; Chen, H.L.; Lin, Y.X.; Hang, C.H.; Yin, H.X.; Shi, J.X. N-Acetylcysteine Suppresses Oxidative Stress in Experimental Rats with Subarachnoid Hemorrhage. J. Clin. Neurosci. 2009, 16, 684–688. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinchese, G.; Cimmino, F.; Cavaliere, G.; Catapano, A.; Fogliano, C.; Lama, A.; Pirozzi, C.; Cristiano, C.; Russo, R.; Petrella, L.; et al. The Hepatic Mitochondrial Alterations Exacerbate Meta-Inflammation in Autism Spectrum Disorders. Antioxidants 2022, 11, 1990. https://doi.org/10.3390/antiox11101990
Trinchese G, Cimmino F, Cavaliere G, Catapano A, Fogliano C, Lama A, Pirozzi C, Cristiano C, Russo R, Petrella L, et al. The Hepatic Mitochondrial Alterations Exacerbate Meta-Inflammation in Autism Spectrum Disorders. Antioxidants. 2022; 11(10):1990. https://doi.org/10.3390/antiox11101990
Chicago/Turabian StyleTrinchese, Giovanna, Fabiano Cimmino, Gina Cavaliere, Angela Catapano, Chiara Fogliano, Adriano Lama, Claudio Pirozzi, Claudia Cristiano, Roberto Russo, Lidia Petrella, and et al. 2022. "The Hepatic Mitochondrial Alterations Exacerbate Meta-Inflammation in Autism Spectrum Disorders" Antioxidants 11, no. 10: 1990. https://doi.org/10.3390/antiox11101990
APA StyleTrinchese, G., Cimmino, F., Cavaliere, G., Catapano, A., Fogliano, C., Lama, A., Pirozzi, C., Cristiano, C., Russo, R., Petrella, L., Meli, R., Mattace Raso, G., Crispino, M., Avallone, B., & Mollica, M. P. (2022). The Hepatic Mitochondrial Alterations Exacerbate Meta-Inflammation in Autism Spectrum Disorders. Antioxidants, 11(10), 1990. https://doi.org/10.3390/antiox11101990