
Enzymatic Epoxidation of Long-Chain Terminal Alkenes by Fungal Peroxygenases

 , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Enzymes
2.2. Model Compounds
2.3. Enzymatic Reactions
2.4. GC-MS Analyses
3. Results and Discussion
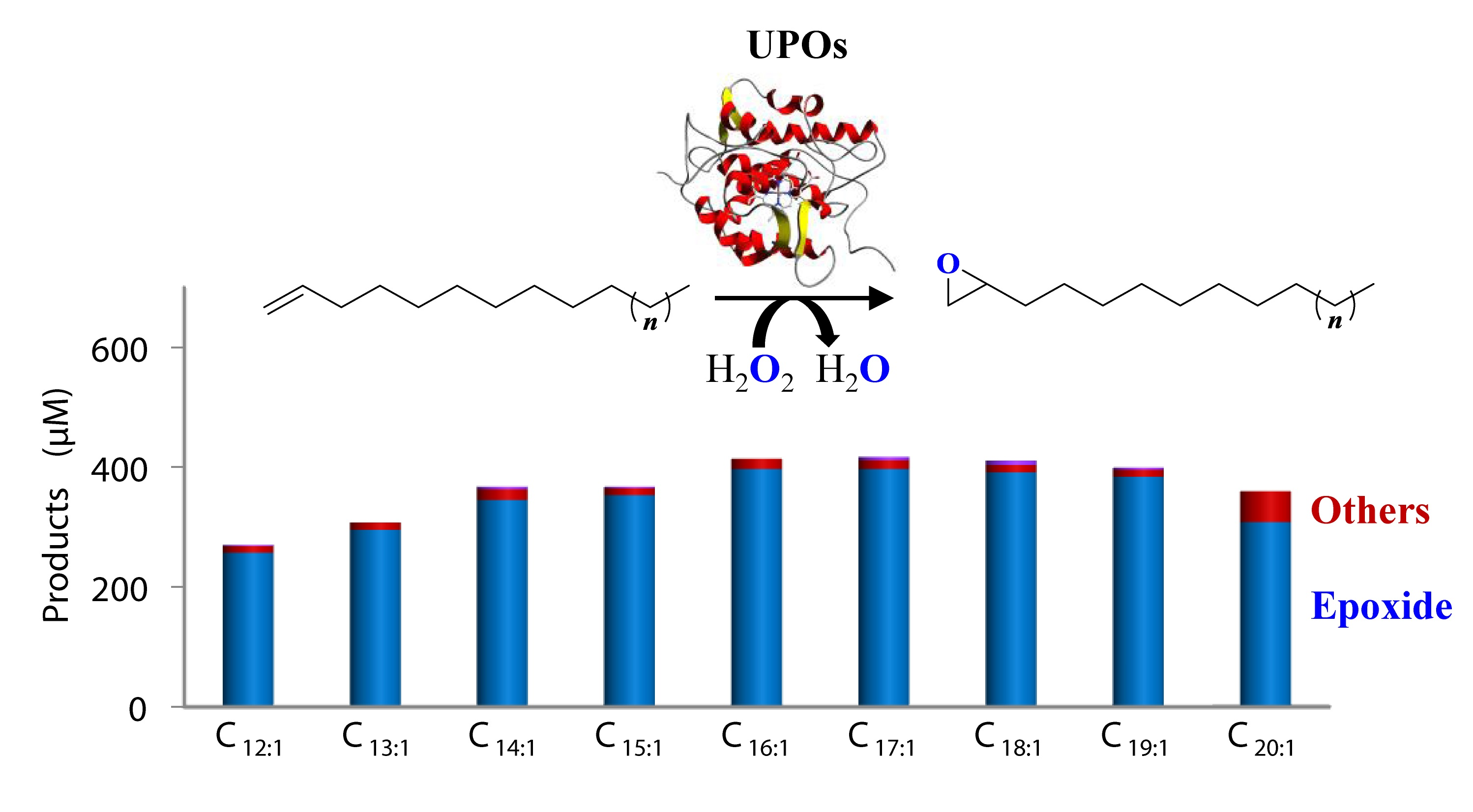
3.1. Optimization of Reaction Conditions (Cosolvent and Cosubstrate) for Conversion of 1-Tetradecene by CglUPO
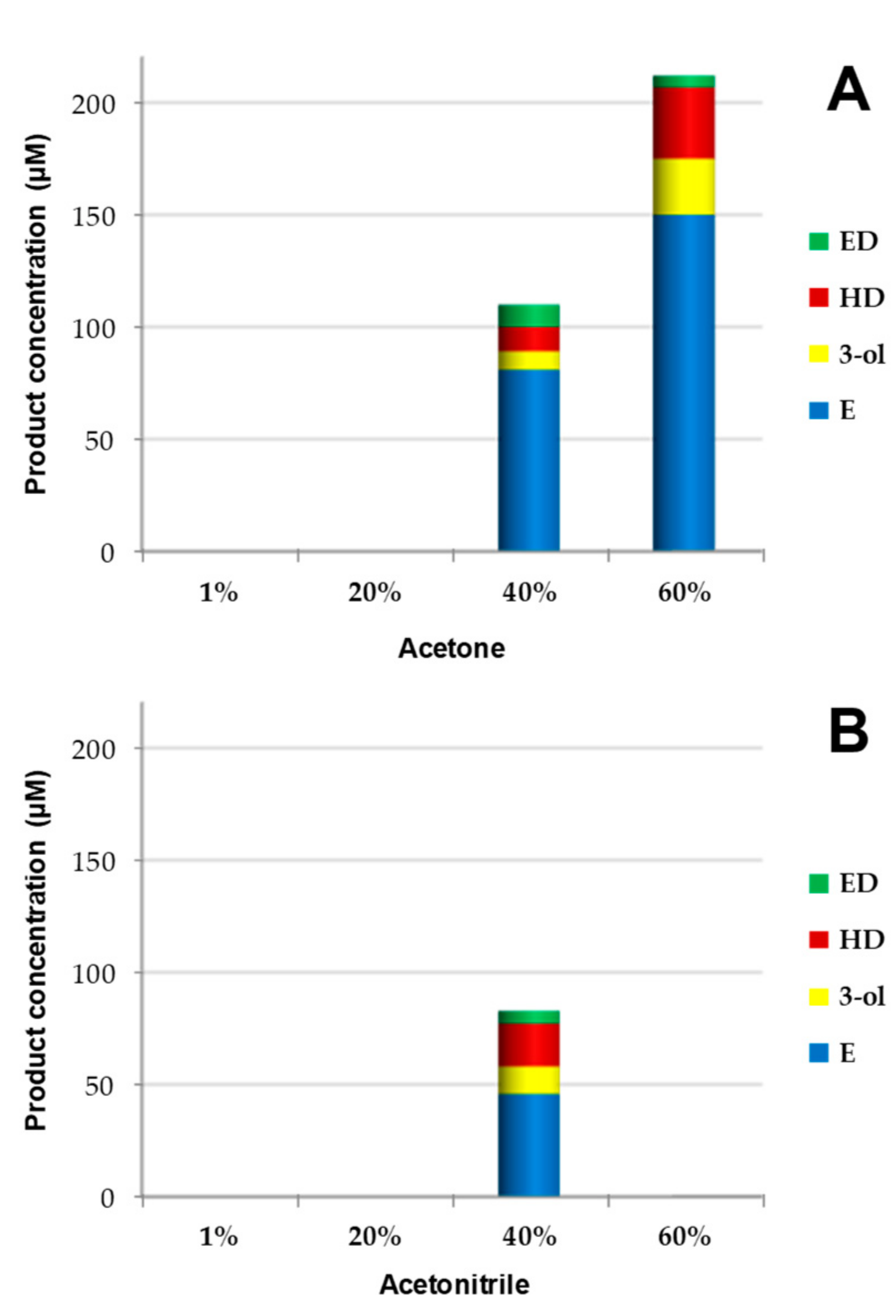
3.2. Selective Epoxidation of 1-Tetradecene by Several UPOs
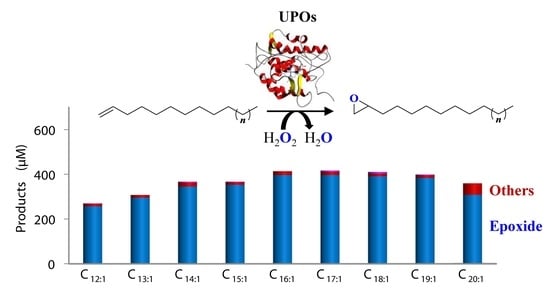
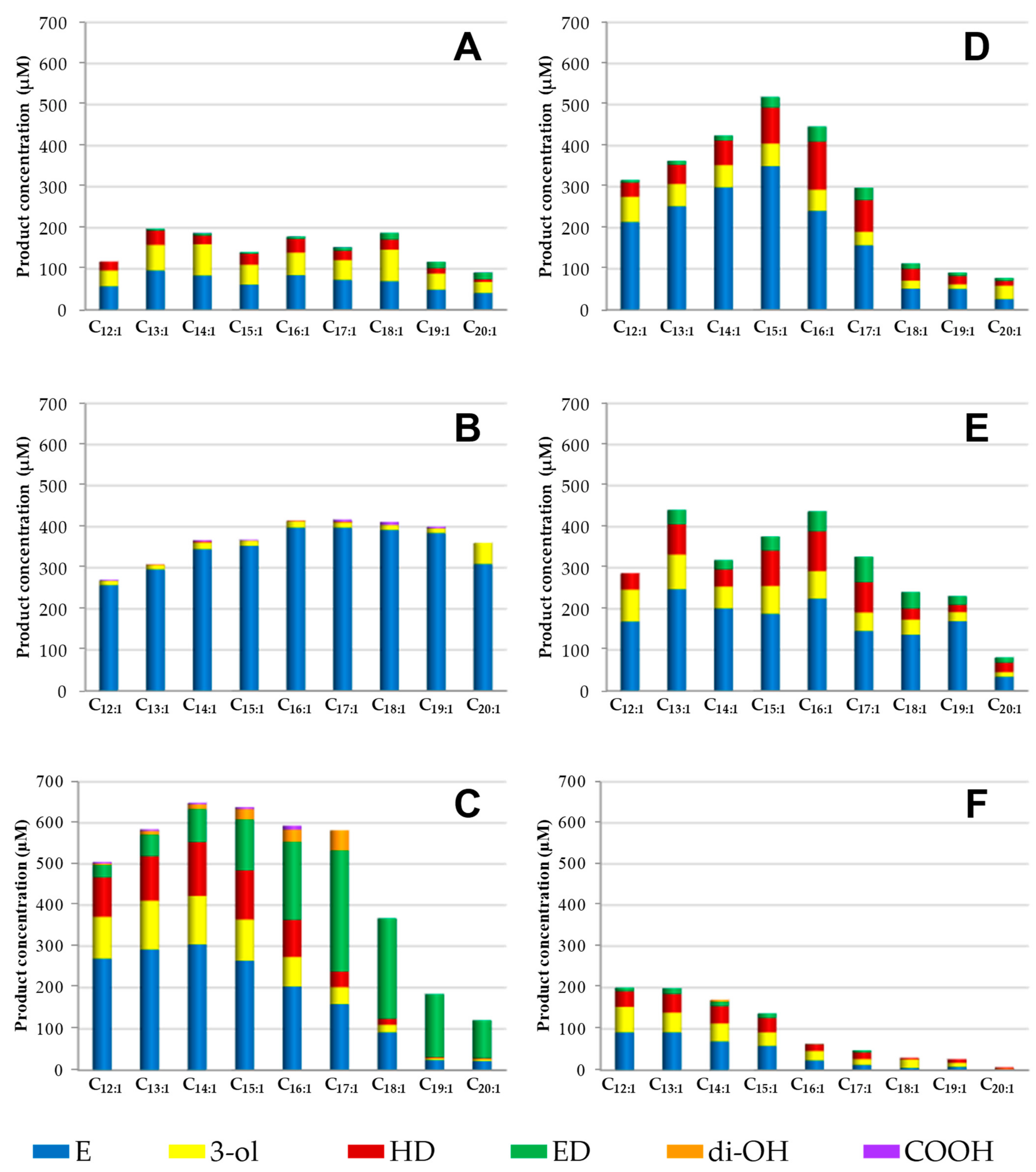
3.3. Selective Epoxidation of Different Long-Chain Alkenes by UPOs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maisonneuve, L.; Lamarzelle, O.; Rix, E.; Grau, E.; Cramail, H. Isocyanate-free routes to polyurethanes and poly(hydroxy urethane)s. Chem. Rev. 2015, 115, 12407–12439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Sena, W.Y.; Cai, X.; Kebir, N.; Vernières-Hassimi, L.; Serra, C.; Salmi, T.; Leveneur, S. Aminolysis of cyclic-carbonate vegetable oils as a non-isocyanate route for the synthesis of polyurethane: A kinetic and thermal study. Chem. Eng. J. 2018, 346, 271–280. [Google Scholar] [CrossRef]
- Maisonneuve, L.; Wirotius, A.L.; Alfos, C.; Grau, E.; Cramail, H. Fatty acid-based (bis) 6-membered cyclic carbonates as efficient isocyanate free poly(hydroxyurethane) precursors. Polym. Chem. 2014, 5, 6142–6147. [Google Scholar] [CrossRef] [Green Version]
- Coombs, J.R.; Morken, J.P. Catalytic enantioselective functionalization of unactivated terminal alkenes. Angew. Chem. Int. Ed. 2016, 55, 2636–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, C.; Tiozzo, C.; Ravasio, N.; Psaro, R.; Carniato, F.; Bisio, C.; Guidotti, M. An efficient epoxidation of terminal aliphatic alkenes over heterogeneous catalysts: When solvent matters. Catal. Sci. Technol. 2016, 6, 3832–3839. [Google Scholar] [CrossRef]
- Köckritz, A.; Martin, A. Oxidation of unsaturated fatty acid derivatives and vegetable oils. Eur. J. Lipid Sci. Technol. 2008, 110, 812–824. [Google Scholar] [CrossRef]
- Wentzel, B.B.; Alsters, P.L.; Feiters, M.C.; Nolte, R.J.M. Mechanistic studies on the Mukaiyama epoxidation. J. Org. Chem. 2004, 69, 3453–3464. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, P.; Zolezzi, S.; Parada, J.; Bunel, E.; Moya, S.A.; Sariego, R. Ruthenium carbonyl complexes in catalytic epoxidation of olefins co-catalyzed by isobutyl-aldehyde. Appl. Organometal. Chem. 2006, 20, 260–263. [Google Scholar] [CrossRef]
- Abraham, M.E.; Benenati, R.F. Kinetics and mechanism of the epoxidation of unsaturated fatty acids. AIChE J. 1972, 18, 807–811. [Google Scholar] [CrossRef]
- Cai, X.; Zheng, J.L.; Aguilera, A.F.; Vernières-Hassimi, L.; Tolvanen, P.; Salmi, T.; Leveneur, S. Influence of ring-opening reactions on the kinetics of cottonseed oil epoxidation. Int. J. Chem. Kinet. 2018, 50, 726–741. [Google Scholar] [CrossRef]
- Pérez-Sena, W.Y.; Wärnå, J.; Eränen, K.; Tolvanen, P.; Estel, L.; Leveneur, S.; Salmi, T. Use of semibatch reactor technology for the investigation of reaction mechanism and kinetics: Heterogeneously catalyzed epoxidation of fatty acid esters. Chem. Eng. Sci. 2021, 230, 116206. [Google Scholar] [CrossRef]
- Zheng, J.L.; Wärnå, J.; Salmi, T.; Burel, F.; Taouk, B.; Leveneur, S. Kinetic modeling strategy for an exothermic multiphase reactor system: Application to vegetable oils epoxidation using Prileschajew method. AIChE J. 2016, 62, 726–741. [Google Scholar] [CrossRef]
- Sato, K.; Aoki, M.; Ogawa, M.; Hashimoto, T.; Noyori, R. A practical method for epoxidation of terminal olefins with 30% hydrogen peroxide under halide-free conditions. J. Org. Chem. 1996, 61, 8310–8311. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Jiang, P.; Wai, P.T.; Gu, Q.; Zhang, W. Recent progress in application of molybdenum-based catalysts for epoxidation of alkenes. Catalysts 2019, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Grigoropoulou, G.; Clark, J.H.; Elings, J.A. Recent developments on the epoxidation of alkenes using hydrogen peroxide as an oxidant. Green Chem. 2003, 5, 1–7. [Google Scholar] [CrossRef]
- Peckh, K.; Lisicki, D.; Talik, G.; Orlinska, B. Oxidation of long-chain a-olefins using environmentally-friendly oxidants. Materials 2020, 13, 4545. [Google Scholar] [CrossRef]
- Xu, Y.; Khaw, N.R.B.J.; Li, Z. Efficient epoxidation of alkenes with hydrogen peroxide, lactone, and lipase. Green Chem. 2009, 11, 2047–2051. [Google Scholar] [CrossRef]
- McClay, K.; Fox, B.G.; Steffan, R.J. Toluene monooxygenase-catalyzed epoxidation of alkenes. Appl. Environ. Microbiol. 2000, 66, 1877–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, A.; Hofstetter, K.; Feiten, H.J.; Hollmann, F.; Witholt, B. Integrated biocatalytic synthesis on gram scale: The highly enantioselective preparation of chiral oxiranes with styrene monooxygenase. Adv. Synth. Catal. 2001, 343, 732–737. [Google Scholar] [CrossRef]
- Elliott, S.J.; Zhu, M.; Tso, L.; Nguyen, H.H.; Yip, J.H.K.; Chan, S.I. Regio- and stereoselectivity of particulate methane monooxygenase from Methylococcus capsulatus (Bath). J. Am. Chem. Soc. 1997, 119, 9949–9955. [Google Scholar] [CrossRef]
- Geigert, J.; Lee, T.D.; Dalietos, D.J.; Hirano, D.S.; Neidleman, S.L. Epoxidation of alkenes by chloroperoxidase catalysis. Biochem. Biophys. Res. Commun. 1986, 136, 778–782. [Google Scholar] [CrossRef]
- Kubo, T.; Peters, M.W.; Meinhold, P.; Arnold, F.H. Enantioselective epoxidation of terminal alkenes to (R)- and (S)-epoxides by engineered cytochromes P450 BM-3. Chem. Eur. J. 2006, 12, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Toda, H.; Ohuchi, T.; Imae, R.; Itoh, N. Microbial production of aliphatic (S)-epoxyalkanes by using Rhodococcus sp. strain ST-10 styrene monooxygenase expressed in organic-solvent-tolerant Kocuria rhizophila DC2201. Appl. Environ. Microbiol. 2015, 81, 1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofrichter, M.; Kellner, H.; Herzog, R.; Karich, A.; Kiebist, J.; Scheibner, K.; Ullrich, R. Peroxide-mediated oxygenation of organic compounds by fungal peroxygenases. Antioxidants 2022, 11, 163. [Google Scholar] [CrossRef]
- Carro, J.; González-Benjumea, A.; Fernández-Fueyo, E.; Aranda, C.; Guallar, V.; Gutiérrez, A.; Martínez, A.T. Modulating fatty acid epoxidation vs hydroxylation in a fungal peroxygenase. ACS Catal. 2019, 9, 6234–6242. [Google Scholar] [CrossRef] [Green Version]
- Municoy, M.; González-Benjumea, A.; Carro, J.; Aranda, C.; Linde, D.; Renau-Mínguez, C.; Ullrich, R.; Hofrichter, M.; Guallar, V.; Gutiérrez, A.; et al. Fatty-acid oxygenation by fungal peroxygenases: From computational simulations to preparative regio- and stereo-selective epoxidation. ACS Catal. 2020, 10, 13584–13595. [Google Scholar] [CrossRef]
- Aranda, C.; Carro, J.; González-Benjumea, A.; Babot, E.D.; Olmedo, A.; Linde, D.; Martínez, A.T.; Gutiérrez, A. Advances in enzymatic oxyfunctionalization of aliphatic compounds. Biotechnol. Adv. 2021, 51, 107703. [Google Scholar] [CrossRef]
- Hofrichter, M.; Ullrich, R.; Pecyna, M.J.; Liers, C.; Lundell, T. New and classic families of secreted fungal heme peroxidases. Appl. Microbiol. Biotechnol. 2010, 87, 871–897. [Google Scholar] [CrossRef] [PubMed]
- Aranda, C.; Ullrich, R.; Kiebist, J.; Scheibner, K.; del Río, J.C.; Hofrichter, M.; Martínez, A.T.; Gutiérrez, A. Selective synthesis of the resveratrol analogue 4,4′-dihydroxy-trans-stilbene and stilbenoids modification by fungal peroxygenases. Catal. Sci. Technol. 2018, 8, 2394–2401. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, A.; Babot, E.D.; Ullrich, R.; Hofrichter, M.; Martínez, A.T.; del Río, J.C. Regioselective oxygenation of fatty acids, fatty alcohols and other aliphatic compounds by a basidiomycete heme-thiolate peroxidase. Arch. Biochem. Biophys. 2011, 514, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Peter, S.; Kinne, M.; Wang, X.; Ulrich, R.; Kayser, G.; Groves, J.T.; Hofrichter, M. Selective hydroxylation of alkanes by an extracellular fungal peroxygenase. FEBS J. 2011, 278, 3667–3675. [Google Scholar] [CrossRef]
- Babot, E.D.; del Río, J.C.; Kalum, L.; Martínez, A.T.; Gutiérrez, A. Oxyfunctionalization of aliphatic compounds by a recombinant peroxygenase from Coprinopsis cinerea. Biotechnol. Bioeng. 2013, 110, 2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, S.; Kinne, M.; Ullrich, R.; Kayser, G.; Hofrichter, M. Epoxidation of linear, branched and cyclic alkenes catalyzed by unspecific peroxygenase. Enzym. Microb. Technol. 2013, 52, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Olmedo, A.; Aranda, C.; del Río, J.C.; Kiebist, J.; Scheibner, K.; Martínez, A.T.; Gutiérrez, A. From alkanes to carboxylic acids: Terminal oxygenation by a fungal peroxygenase. Angew. Chem. Int. Ed. 2016, 55, 12248–12251. [Google Scholar] [CrossRef]
- Aranda, C.; Olmedo, A.; Kiebist, J.; Scheibner, K.; del Río, J.C.; Martínez, A.T.; Gutiérrez, A. Selective epoxidation of fatty acids and fatty acid methyl esters by fungal peroxygenases. ChemCatChem 2018, 10, 3964–3968. [Google Scholar] [CrossRef] [Green Version]
- González-Benjumea, A.; Carro, J.; Renau, C.; Linde, D.; Fernández-Fueyo, E.; Gutiérrez, A.; Martínez, A.T. Fatty acid epoxidation by Collariella virescens peroxygenase and heme-channel variants. Catal. Sci. Technol. 2020, 10, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Linde, D.; Olmedo, A.; González-Benjumea, A.; Renau, C.; Estévez, M.; Carro, J.; Fernández-Fueyo, E.; Gutiérrez, A.; Martínez, A.T. Two new unspecific peroxygenases from heterologous expression of fungal genes in Escherichia coli. Appl. Environ. Microbiol. 2020, 86, e02899-19. [Google Scholar] [CrossRef] [Green Version]
- Olmedo, A.; del Río, J.C.; Kiebist, J.; Scheibner, K.; Martínez, A.T.; Gutiérrez, A. Fatty acid chain shortening by a fungal peroxygenase. Chem. Eur. J. 2017, 23, 16985–16989. [Google Scholar] [CrossRef] [Green Version]
- Babot, E.D.; del Río, J.C.; Cañellas, M.; Sancho, F.; Lucas, F.; Guallar, V.; Kalum, L.; Lund, H.; Gröbe, G.; Scheibner, K.; et al. Steroid hydroxylation by basidiomycete peroxygenases: A combined experimental and computational study. Appl. Environ. Microbiol. 2015, 81, 4130–4142. [Google Scholar] [CrossRef] [Green Version]
- Kiebist, J.; Schmidtke, K.U.; Zimmermann, J.; Kellner, H.; Jehmlich, N.; Ullrich, R.; Zänder, D.; Hofrichter, M.; Scheibner, K. A peroxygenase from Chaetomium globosum catalyzes the selective oxygenation of testosterone. ChemBioChem 2017, 18, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Babot, E.D.; del Río, J.C.; Kalum, L.; Martínez, A.T.; Gutiérrez, A. Regioselective hydroxylation in the production of 25-hydroxyvitamin D by Coprinopsis cinerea peroxygenase. ChemCatChem 2015, 7, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Lucas, F.; Babot, E.D.; del Río, J.C.; Kalum, L.; Ullrich, R.; Hofrichter, M.; Guallar, V.; Martínez, A.T.; Gutiérrez, A. Molecular determinants for selective C25-hydroxylation of vitamins D2 and D3 by fungal peroxygenases. Catal. Sci. Technol. 2016, 6, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Aranda, C.; Municoy, M.; Guallar, V.; Kiebist, J.; Scheibner, K.; Ullrich, R.; del Río, J.C.; Hofrichter, M.; Martínez, A.T.; Gutiérrez, A. Selective synthesis of 4-hydroxyisophorone and 4-ketoisophorone by fungal peroxygenases. Catal. Sci. Technol. 2019, 9, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Babot, E.D.; Aranda, C.; del Río, J.C.; Ullrich, R.; Kiebist, J.; Scheibner, K.; Hofrichter, M.; Martínez, A.T.; Gutiérrez, A. Selective oxygenation of ionones and damascones by fungal peroxygenases. J. Agric. Food Chem. 2020, 68, 5375–5383. [Google Scholar] [CrossRef]
- Ullrich, R.; Nuske, J.; Scheibner, K.; Spantzel, J.; Hofrichter, M. Novel haloperoxidase from the agaric basidiomycete Agrocybe aegerita oxidizes aryl alcohols and aldehydes. Appl. Environ. Microbiol. 2004, 70, 4575–4581. [Google Scholar] [CrossRef] [Green Version]
- Anh, D.H.; Ullrich, R.; Benndorf, D.; Svatos, A.; Muck, A.; Hofrichter, M. The coprophilous mushroom Coprinus radians secretes a haloperoxidase that catalyzes aromatic peroxygenation. Appl. Environ. Microbiol. 2007, 73, 5477–5485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gröbe, G.; Ullrich, M.; Pecyna, M.; Kapturska, D.; Friedrich, S.; Hofrichter, M.; Scheibner, K. High-yield production of aromatic peroxygenase by the agaric fungus Marasmius rotula. AMB Express 2011, 1, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Landvick, S.; Ostergaard, L.H.; Kalum, L. Polypeptides Having Peroxygenase Activity. International Patent WO2014056916A3, 17 April 2014. [Google Scholar]
- Collman, J.P.; Hampton, P.D.; Brauman, J.T. Suicide inactivation of cytochrome P-450 model compounds by terminal olefins. 1. A mechanistic study of heme N-alkylation and epoxidation. J. Am. Chem. Soc. 1990, 112, 2977–2986. [Google Scholar] [CrossRef]
- Lund, H.; Kalum, L.; Hofrichter, M.; Peter, S. Epoxidation Using Peroxygenase. U.S. Patent US9908860B2, 6 March 2018. [Google Scholar]
- González-Benjumea, A.; Marques, G.; Herold-Majumdar, O.M.; Kiebist, J.; Scheibner, K.; del Río, J.C.; Martínez, A.T.; Gutiérrez, A. High epoxidation yields of vegetable oil hydrolyzates and methyl esters by selected fungal peroxygenases. Front. Bioeng. Biotechnol. 2021, 8, 605854. [Google Scholar] [CrossRef] [PubMed]
- Pecyna, M.J.; Schnorr, K.M.; Ullrich, R.; Scheibner, K.; Kluge, M.G.; Hofrichter, M. Fungal Peroxygenases and Methods of Application. International Patent WO2008/119780A2, 9 October 2008. [Google Scholar]
- Otey, C.R. High-throughput carbon monoxide binding assay for cytochromes P450. Methods Mol. Biol. 2003, 230, 137–139. [Google Scholar]
- Meng, Y.; Taddeo, F.; Aguilera, A.F.; Cai, X.; Russo, V.; Tolvanen, P.; Leveneur, S. The Lord of the chemical rings: Catalytic synthesis of important industrial epoxide compounds. Catalysts 2021, 11, 765. [Google Scholar] [CrossRef]
- Karich, A.; Scheibner, K.; Ullrich, R.; Hofrichter, M. Exploring the catalase activity of unspecific peroxygenases and the mechanism of peroxide-dependent heme destruction. J. Mol. Catal. B Enzym. 2016, 134, 238–246. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
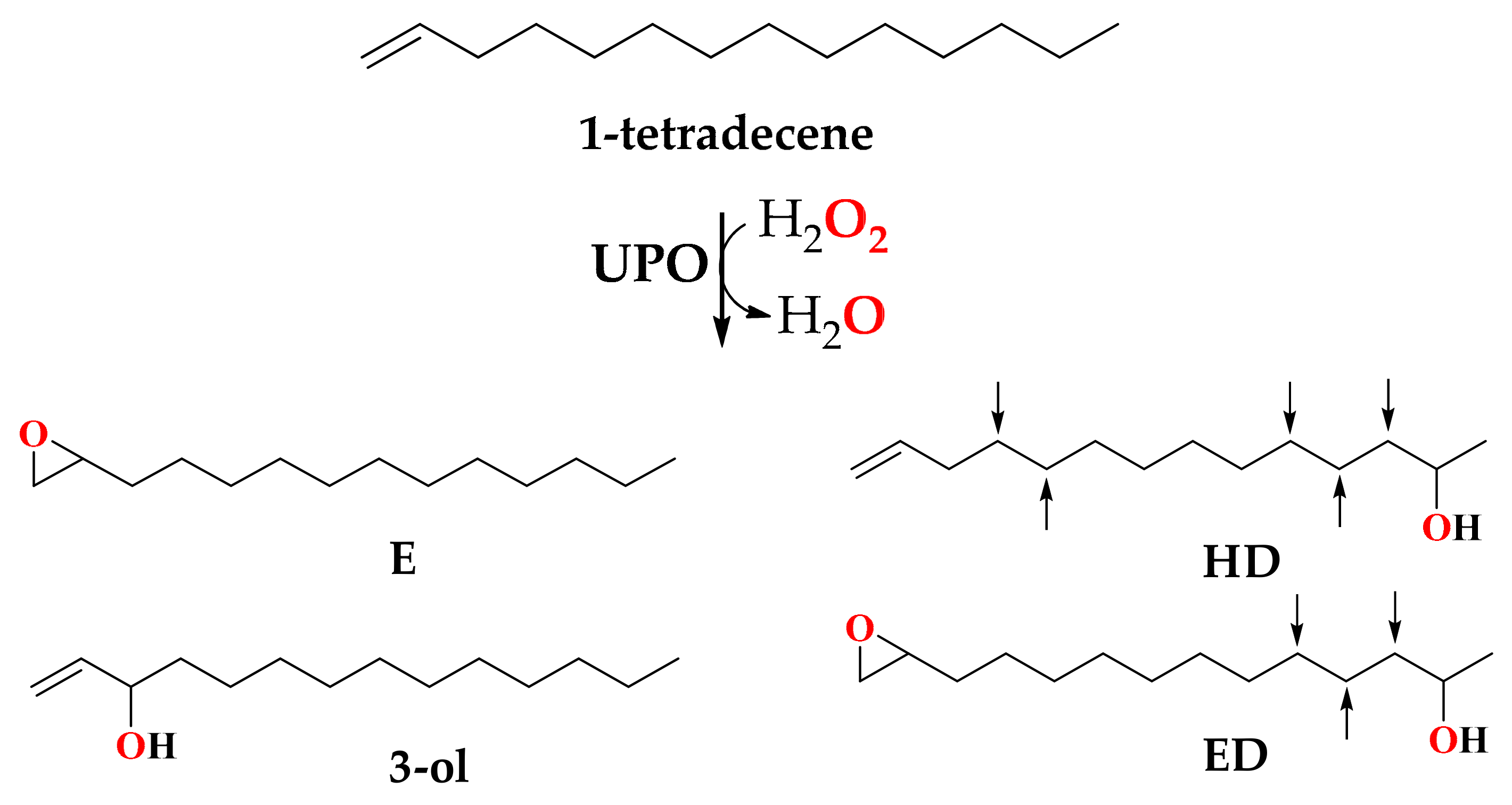{kind=link}
| Enzyme (µM) | Cosubstrate (Type) | Cosubstrate (mM) | Total (µM) | Products (%) | |||
|---|---|---|---|---|---|---|---|
| E | 3-ol | HD | ED | ||||
| 0.5 | H2O2 | 0.5 | 11 | 83 | 17 | 0 | 0 |
| 0.5 | H2O2 | 1 | 51 | 64 | 13 | 24 | 0 |
| 1 | H2O2 | 1 | 49 | 68 | 9 | 13 | 10 |
| 1 | H2O2 | 5 | 46 | 63 | 10 | 23 | 4 |
| 2 | H2O2 | 0.5 | 100 | 71 | 12 | 14 | 4 |
| 2 | H2O2 | 1 | 195 | 72 | 12 | 14 | 2 |
| 2 | H2O2 | 5 | 134 | 73 | 12 | 14 | 2 |
| 2 | tBuOOH | 0.5 | 91 | 69 | 11 | 15 | 5 |
| 2 | tBuOOH | 1 | 94 | 68 | 12 | 17 | 3 |
| 2 | tBuOOH | 5 | 28 | 82 | 18 | 0 | 0 |
| Enzyme (µM) | pH | Total (µM) | Products (%) | |||
|---|---|---|---|---|---|---|
| E | 3-ol | HD | ED | |||
| AaeUPO | 7.0 | 127 | 60 | 21 | 16 | 3 |
| 5.5 | 110 | 61 | 21 | 18 | 0 | |
| MroUPO | 7.0 | 89 | 95 | 5 | - | - |
| 5.5 | 137 | 96 | 4 | - | - | |
| rCciUPO | 7.0 | 265 | 43 | 19 | 26 | 12 |
| 5.5 | 317 | 44 | 18 | 27 | 11 | |
| CglUPO | 7.0 | 212 | 71 | 12 | 15 | 2 |
| 5.5 | 164 | 70 | 11 | 14 | 5 | |
| rHinUPO | 7.0 | 210 | 58 | 17 | 22 | 3 |
| 5.5 | 271 | 58 | 16 | 23 | 3 | |
| rDcaUPO | 7.0 | 160 | 44 | 22 | 28 | 4 |
| 5.5 | 180 | 45 | 20 | 27 | 8 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babot, E.D.; Aranda, C.; Kiebist, J.; Scheibner, K.; Ullrich, R.; Hofrichter, M.; Martínez, A.T.; Gutiérrez, A. Enzymatic Epoxidation of Long-Chain Terminal Alkenes by Fungal Peroxygenases. Antioxidants 2022, 11, 522. https://doi.org/10.3390/antiox11030522
Babot ED, Aranda C, Kiebist J, Scheibner K, Ullrich R, Hofrichter M, Martínez AT, Gutiérrez A. Enzymatic Epoxidation of Long-Chain Terminal Alkenes by Fungal Peroxygenases. Antioxidants. 2022; 11(3):522. https://doi.org/10.3390/antiox11030522
Chicago/Turabian StyleBabot, Esteban D., Carmen Aranda, Jan Kiebist, Katrin Scheibner, René Ullrich, Martin Hofrichter, Angel T. Martínez, and Ana Gutiérrez. 2022. "Enzymatic Epoxidation of Long-Chain Terminal Alkenes by Fungal Peroxygenases" Antioxidants 11, no. 3: 522. https://doi.org/10.3390/antiox11030522
APA StyleBabot, E. D., Aranda, C., Kiebist, J., Scheibner, K., Ullrich, R., Hofrichter, M., Martínez, A. T., & Gutiérrez, A. (2022). Enzymatic Epoxidation of Long-Chain Terminal Alkenes by Fungal Peroxygenases. Antioxidants, 11(3), 522. https://doi.org/10.3390/antiox11030522