
Activation of AMPK/miR-181b Axis Alleviates Endothelial Dysfunction and Vascular Inflammation in Diabetic Mice


 , , ,
, , , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Arteries
2.2. Animal Experimentation
2.3. Blood Glucose Measurement
2.4. Organ Culture of Arteries
2.5. Functional Assay by Wire Myography
2.6. Lucigenin-Enhanced Chemiluminescence Assay
2.7. ROS Determination by Dihydroethidium (DHE) Staining
2.8. Cell Culture
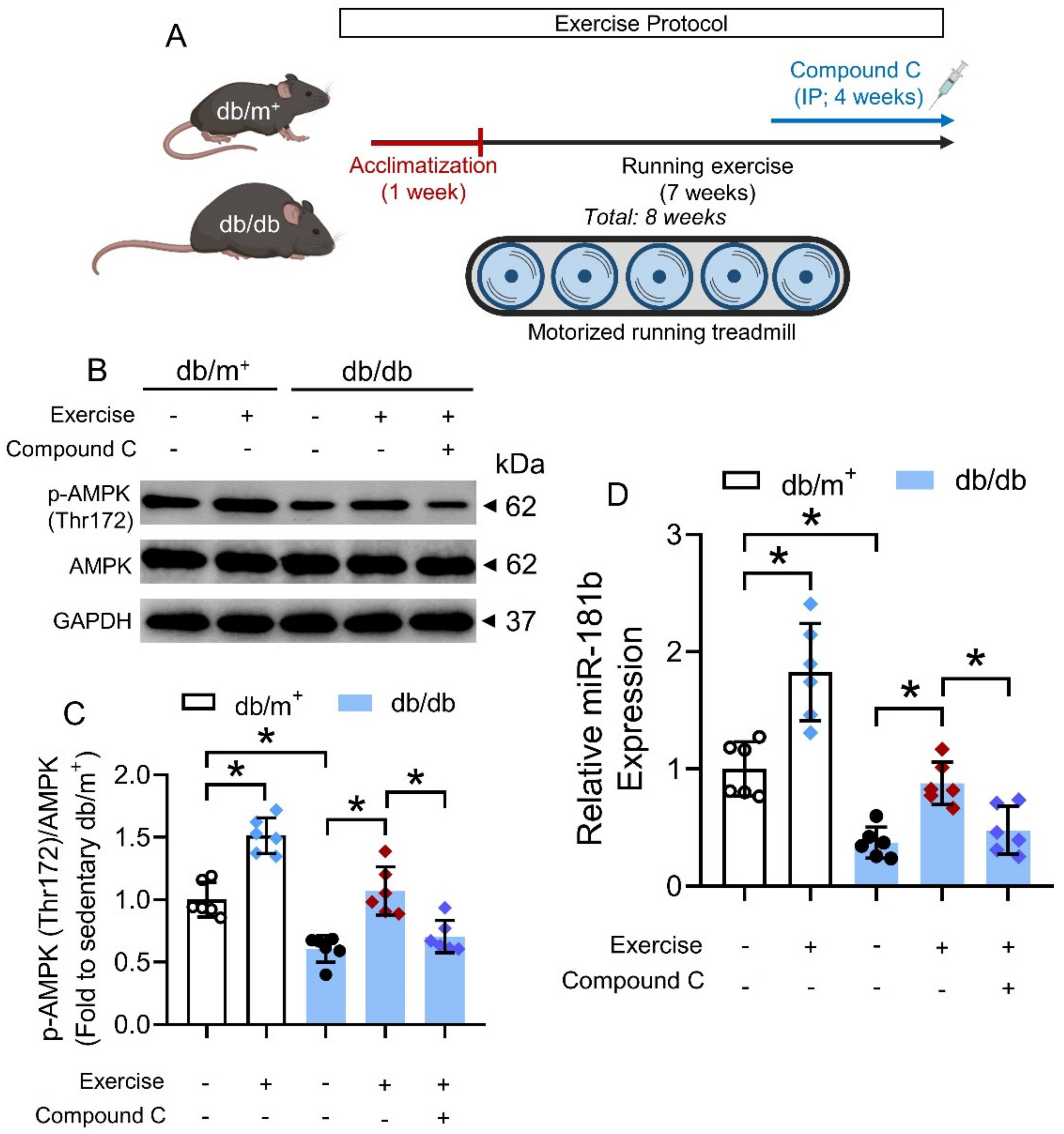
2.9. Exercise Protocol
2.10. Hemodynamic Study
2.11. Quantitative RT-PCR
2.12. Western Blotting
2.13. Experimental Blinding and Randomization
2.14. Statistical Analysis
3. Results
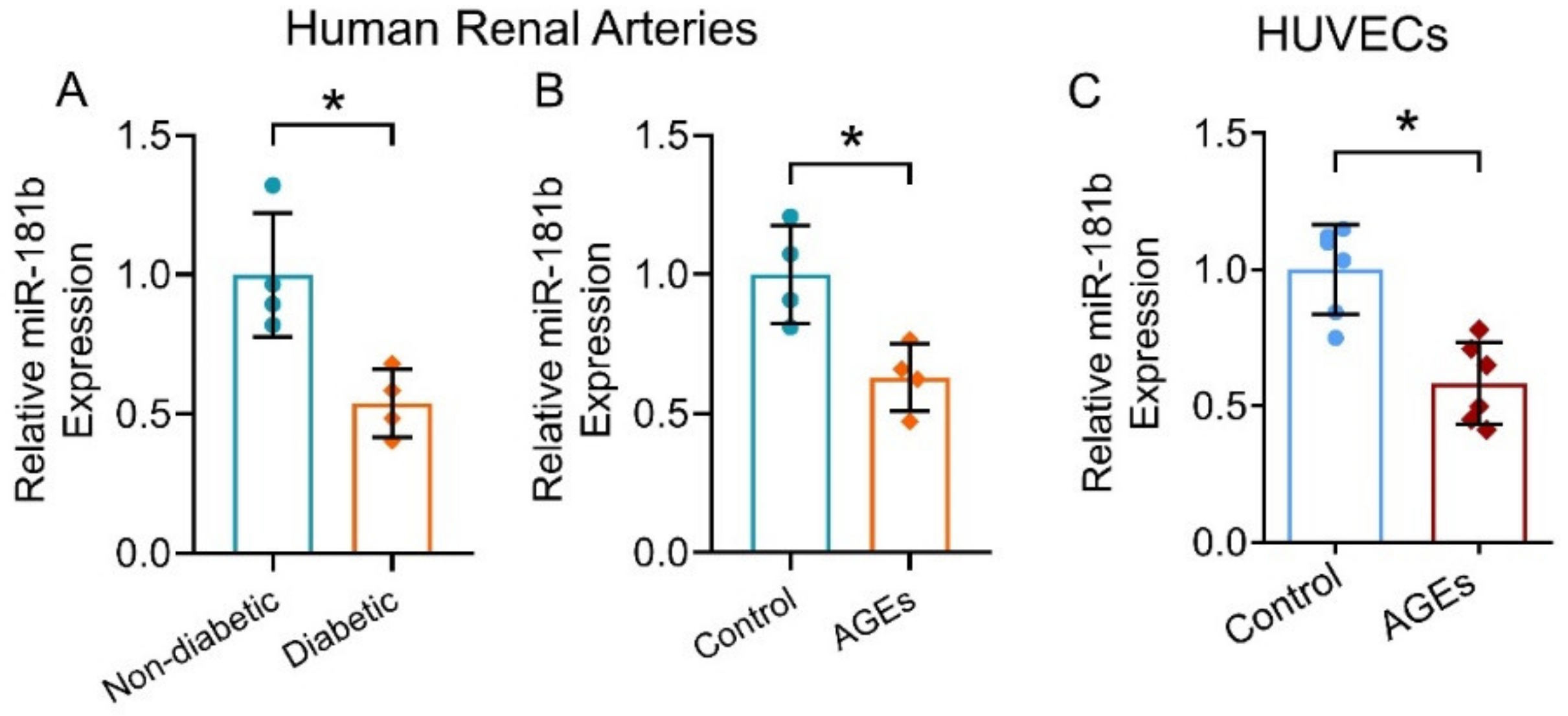
3.1. miR-181b Expression Reduced in Diabetic Condition
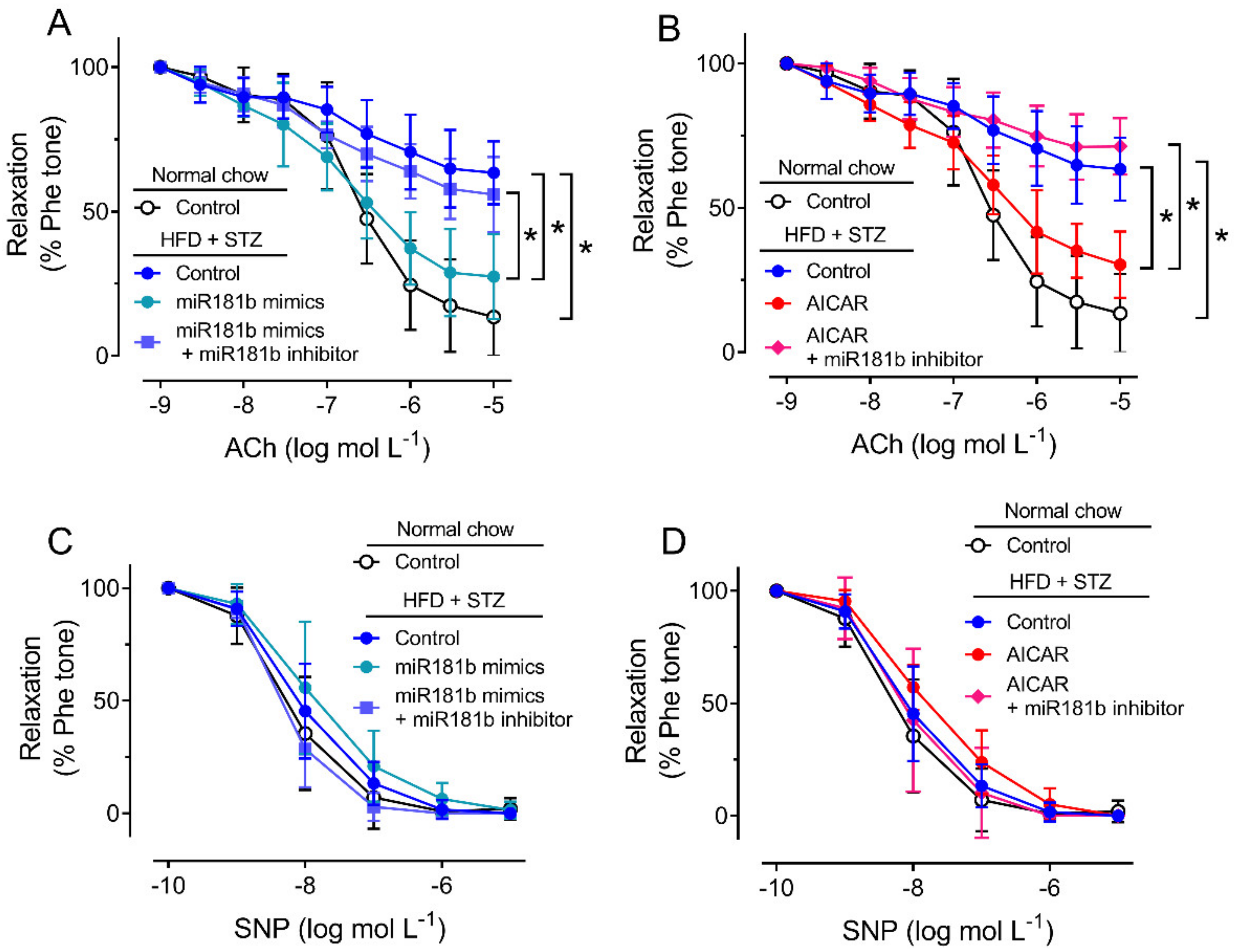
3.2. miR-181b Overexpression Improved Endothelial Function in Aortas of Diabetic Mice
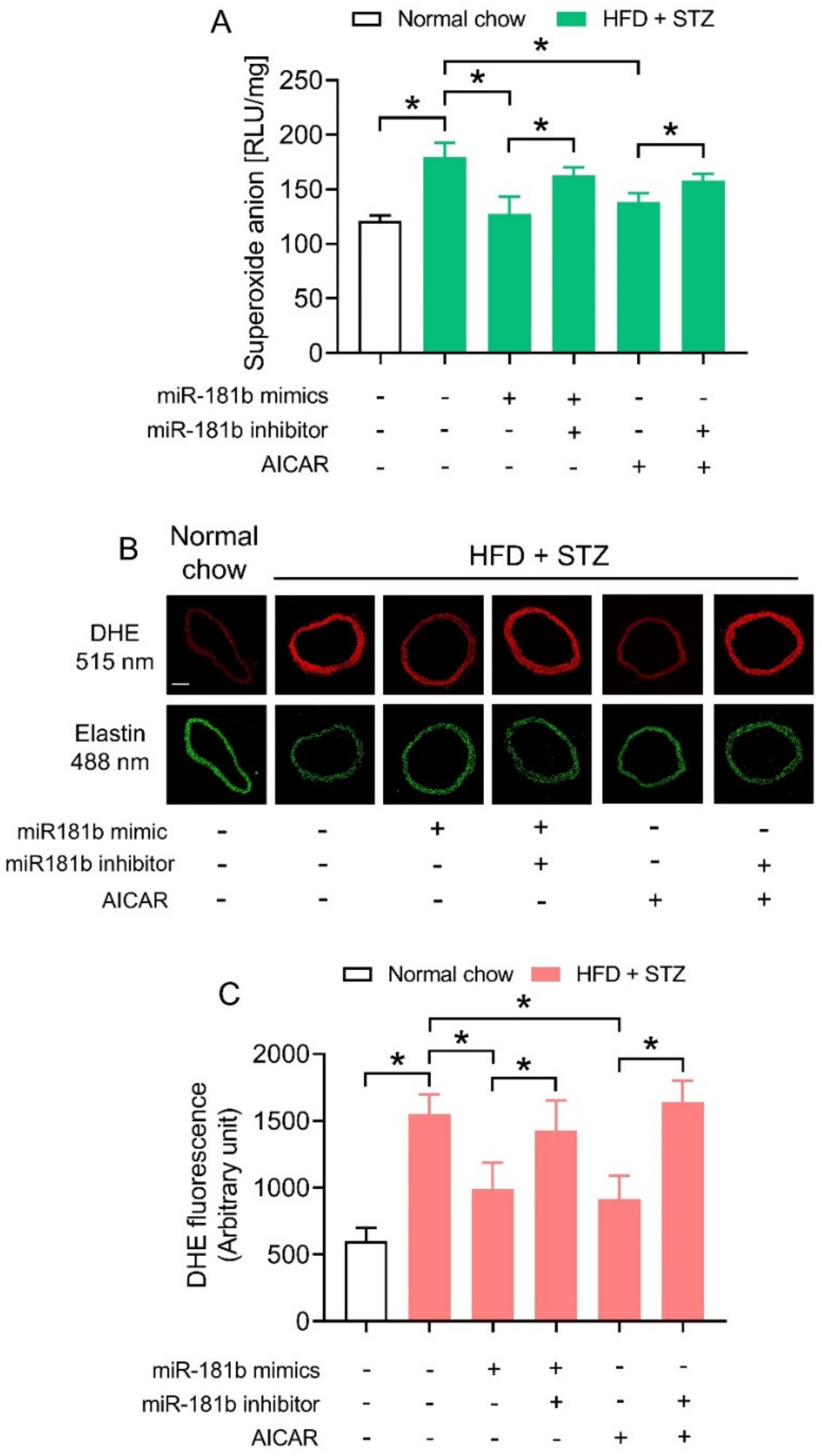
3.3. miR-181b Overexpression Suppressed Vascular ROS in Aortas of Diabetic Mice
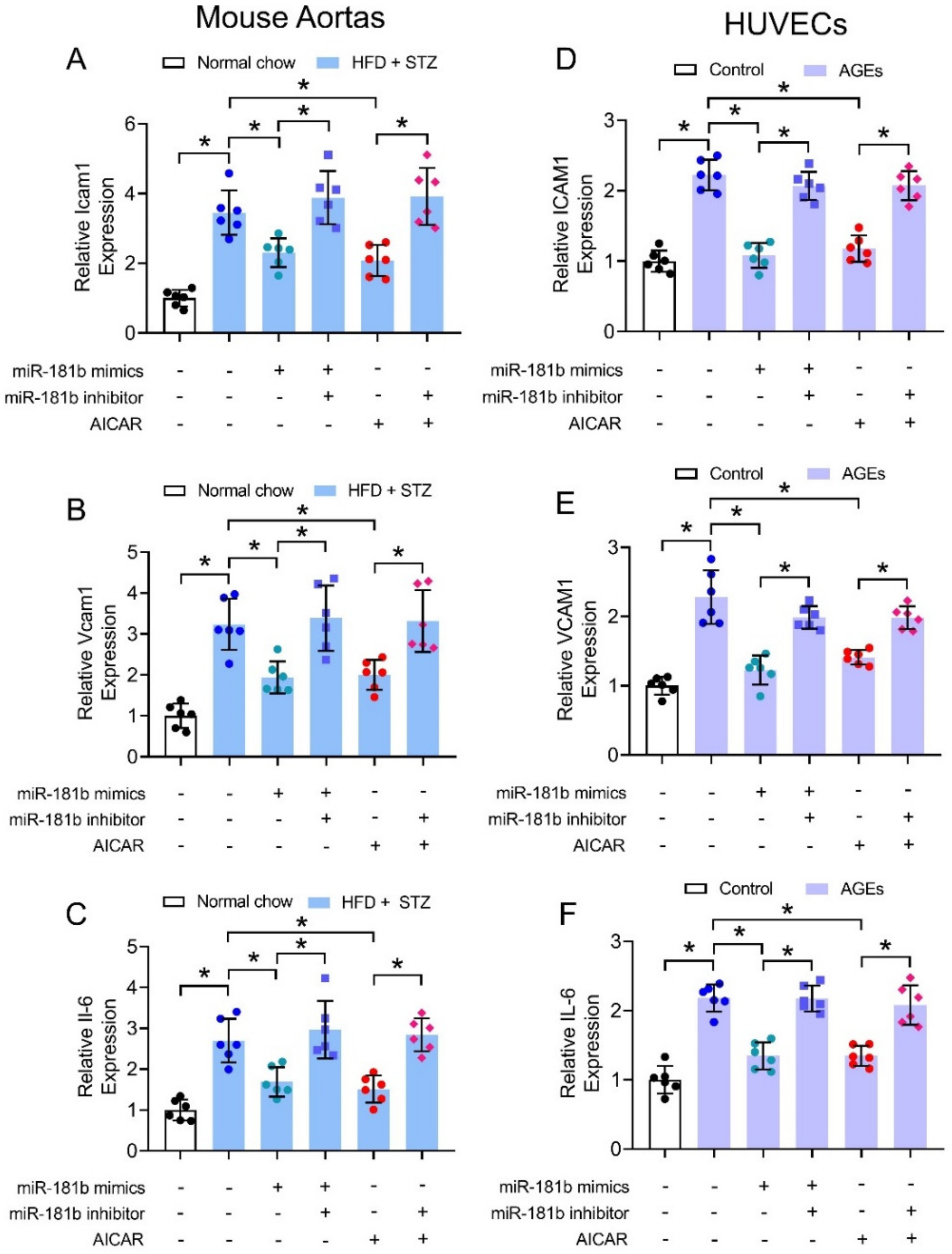
3.4. miR-181b Overexpression Inhibited Vascular and Endothelial Inflammation
3.5. AMPK Activation Increased miR-181b Expression in Human Arteries and Endothelial Cells
3.6. Chronic Exercise Activated AMPK/miR-181b Axis in Diabetic Mice
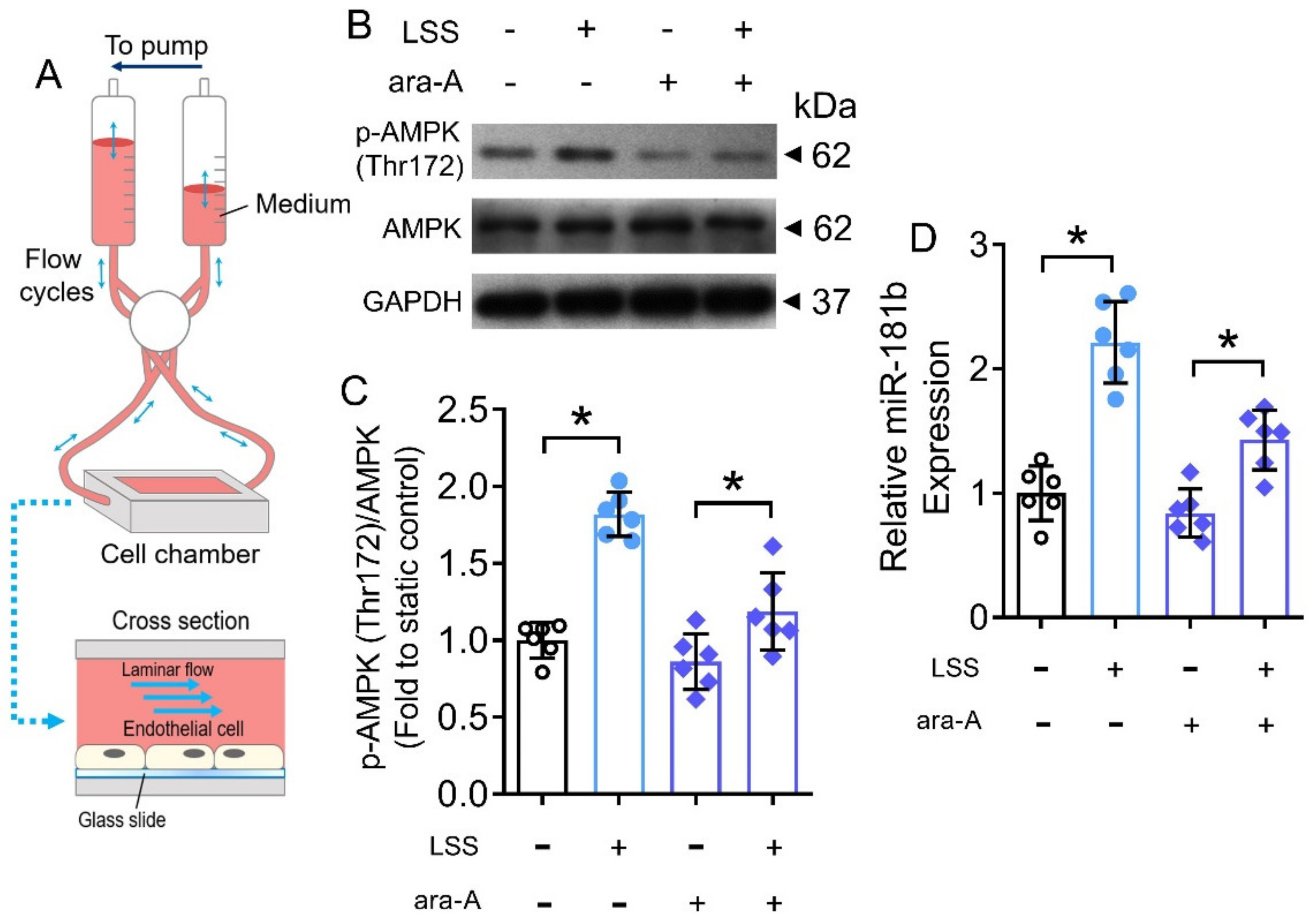
3.7. LSS Activated AMPK/miR-181b Axis in Endothelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ling, W.; Huang, Y.; Huang, Y.M.; Fan, R.R.; Sui, Y.; Zhao, H.L. Global Trend of Diabetes Mortality Attributed to Vascular Complications, 2000–2016. Cardiovasc. Diabetol. 2020, 19, 182. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 Diabetes Mellitus. Nat. Rev. Dis. Prim. 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- Meza, C.A.; LaFavor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Harrison, D.G. Endothelial Dysfunction in Cardiovascular Diseases: The Role of Oxidant Stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, A.O.; Jacobs, D.R.; Sanchez, O.A.; Goff, D.C.; Reiner, A.P.; Gross, M.D. Oxidative Stress, Inflammation, Endothelial Dysfunction and Incidence of Type 2 Diabetes. Cardiovasc. Diabetol. 2016, 15, 51. [Google Scholar] [CrossRef] [Green Version]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.T.B.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cheng, C.K.; Yi, M.; Lui, K.O.; Huang, Y. Targeting Endothelial Dysfunction and Inflammation. J. Mol. Cell. Cardiol. 2022, 168, 58–67. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; MacRae, I.J. Regulation of MicroRNA Function in Animals. Nat. Rev. Mol. Cell Biol. 2018, 20, 21–37. [Google Scholar] [CrossRef]
- Pescador, N.; Pérez-Barba, M.; Ibarra, J.M.; Corbatón, A.; Martínez-Larrad, M.T.; Serrano-Ríos, M. Serum Circulating MicroRNA Profiling for Identification of Potential Type 2 Diabetes and Obesity Biomarkers. PLoS ONE 2013, 8, e77251. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, M.; Witkowski, M.; Saffarzadeh, M.; Friebel, J.; Tabaraie, T.; Ta Bao, L.; Chakraborty, A.; Dörner, A.; Stratmann, B.; Tschoepe, D.; et al. Vascular MiR-181b Controls Tissue Factor-Dependent Thrombogenicity and Inflammation in Type 2 Diabetes. Cardiovasc. Diabetol. 2020, 19, 20. [Google Scholar] [CrossRef]
- Sun, X.; Icli, B.; Wara, A.K.; Belkin, N.; He, S.; Kobzik, L.; Hunninghake, G.M.; Vera, M.P.; Blackwell, T.S.; Baron, R.M.; et al. MicroRNA-181b Regulates NF-ΚB–Mediated Vascular Inflammation. J. Clin. Investig. 2012, 122, 1973. [Google Scholar] [CrossRef]
- Sun, X.; He, S.; Wara, A.K.M.; Icli, B.; Shvartz, E.; Tesmenitsky, Y.; Belkin, N.; Li, D.; Blackwell, T.S.; Sukhova, G.K.; et al. Systemic Delivery of MicroRNA-181b Inhibits Nuclear Factor-ΚB Activation, Vascular Inflammation, and Atherosclerosis in Apolipoprotein E-Deficient Mice. Circ. Res. 2014, 114, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2017, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Cheang, W.S.; Tian, X.Y.; Wong, W.T.; Lau, C.W.; Lee, S.S.T.; Chen, Z.Y.; Yao, X.; Wang, N.; Huang, Y. Metformin Protects Endothelial Function in Diet-Induced Obese Mice by Inhibition of Endoplasmic Reticulum Stress through 5’ Adenosine Monophosphate-Activated Protein Kinase-Peroxisome Proliferator-Activated Receptor δ Pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 830–836. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Song, A.; Hu, W.; Dai, M. The Anti-Atherosclerotic Effect of Paeonol against Vascular Smooth Muscle Cell Proliferation by Up-Regulation of Autophagy via the AMPK/MTOR Signaling Pathway. Front. Pharmacol. 2018, 8, 948. [Google Scholar] [CrossRef] [Green Version]
- Gélinas, R.; Mailleux, F.; Dontaine, J.; Bultot, L.; Demeulder, B.; Ginion, A.; Daskalopoulos, E.P.; Esfahani, H.; Dubois-Deruy, E.; Lauzier, B.; et al. AMPK Activation Counteracts Cardiac Hypertrophy by Reducing O-GlcNAcylation. Nat. Commun. 2018, 9, 374. [Google Scholar] [CrossRef] [Green Version]
- Ait-Aissa, K.; Nguyen, Q.M.; Gabani, M.; Kassan, A.; Kumar, S.; Choi, S.K.; Gonzalez, A.A.; Khataei, T.; Sahyoun, A.M.; Chen, C.; et al. MicroRNAs and Obesity-Induced Endothelial Dysfunction: Key Paradigms in Molecular Therapy. Cardiovasc. Diabetol. 2020, 19, 136. [Google Scholar] [CrossRef]
- Gongol, B.; Marin, T.; Zhang, J.; Wang, S.C.; Sun, W.; He, M.; Chen, S.; Chen, L.; Li, J.; Liu, J.H.; et al. Shear Stress Regulation of MiR-93 and MiR-484 Maturation through Nucleolin. Proc. Natl. Acad. Sci. USA 2019, 116, 12974–12979. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of Shear Stress on Endothelial Cells: Go with the Flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Huang, J.; Pu, Y.; Zhang, H.; Xie, L.; He, L.; Zhang, C.L.; Cheng, C.K.; Huo, Y.; Wan, S.; Chen, S.; et al. KLF2 Mediates the Suppressive Effect of Laminar Flow on Vascular Calcification by Inhibiting Endothelial BMP/SMAD1/5 Signaling. Circ. Res. 2021, 129, E87–E100. [Google Scholar] [CrossRef]
- Chiu, J.J.; Chien, S. Effects of Disturbed Flow on Vascular Endothelium: Pathophysiological Basis and Clinical Perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [Green Version]
- Marin, T.; Gongol, B.; Chen, Z.; Woo, B.; Subramaniam, S.; Chien, S.; Shyy, J.Y.J. Mechanosensitive MicroRNAs—Role in Endothelial Responses to Shear Stress and Redox State. Free Radic. Biol. Med. 2013, 64, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Dixit, M.; Bess, E.; Fisslthaler, B.; Härtel, F.V.; Noll, T.; Busse, R.; Fleming, I. Shear Stress-Induced Activation of the AMP-Activated Protein Kinase Regulates FoxO1a and Angiopoietin-2 in Endothelial Cells. Cardiovasc. Res. 2008, 77, 160–168. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Animal Research: Reporting in Vivo Experiments: The ARRIVE Guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. Pharmacol. 2015, 70, 5–47. [Google Scholar] [CrossRef]
- Cheng, C.K.; Wang, C.; Shang, W.; Lau, C.W.; Luo, J.Y.; Wang, L.; Huang, Y. A High Methionine and Low Folate Diet Alters Glucose Homeostasis and Gut Microbiome. Biochem. Biophys. Rep. 2021, 25, 100921. [Google Scholar] [CrossRef]
- Li, Q.; Kim, Y.R.; Vikram, A.; Kumar, S.; Kassan, M.; Gabani, M.; Lee, S.K.; Jacobs, J.S.; Irani, K. P66Shc-Induced MicroRNA-34a Causes Diabetic Endothelial Dysfunction by Downregulating Sirtuin1. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2394–2403. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.K.; Luo, J.Y.; Lau, C.W.; Cho, W.C.S.; Ng, C.F.; Ma, R.C.W.; Tian, X.Y.; Huang, Y. A GLP-1 Analog Lowers ER Stress and Enhances Protein Folding to Ameliorate Homocysteine-Induced Endothelial Dysfunction. Acta Pharmacol. Sin. 2021, 42, 1598–1609. [Google Scholar] [CrossRef]
- Choy, K.W.; Mustafa, M.R.; Lau, Y.S.; Liu, J.; Murugan, D.; Lau, C.W.; Wang, L.; Zhao, L.; Huang, Y. Paeonol Protects against Endoplasmic Reticulum Stress-Induced Endothelial Dysfunction via AMPK/PPARδ Signaling Pathway. Biochem. Pharmacol. 2016, 116, 51–62. [Google Scholar] [CrossRef]
- Ling, W.C.; Liu, J.; Lau, C.W.; Murugan, D.D.; Mustafa, M.R.; Huang, Y. Treatment with Salvianolic Acid B Restores Endothelial Function in Angiotensin II-Induced Hypertensive Mice. Biochem. Pharmacol. 2017, 136, 76–85. [Google Scholar] [CrossRef]
- Gou, L.; Zhao, L.; Song, W.; Wang, L.; Liu, J.; Zhang, H.; Lau, C.W.; Yao, X.; Tian, X.Y.; Wong, W.T.; et al. Inhibition of MiR-92a Suppresses Oxidative Stress and Improves Endothelial Function by Upregulating Heme Oxygenase-1 in Db/Db Mice. Antioxid. Redox Signal. 2018, 28, 358–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheang, W.S.; Wong, W.T.; Zhao, L.; Xu, J.; Wang, L.; Lau, C.W.; Chen, Z.Y.; Ma, R.C.W.; Xu, A.; Wang, N.; et al. PPARδ Is Required for Exercise to Attenuate Endoplasmic Reticulum Stress and Endothelial Dysfunction in Diabetic Mice. Diabetes 2017, 66, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.K.; Miller, I.; Aja, S.; Landree, L.E.; Pinn, M.; McFadden, J.; Kuhajda, F.P.; Moran, T.H.; Ronnett, G.V. C75, a Fatty Acid Synthase Inhibitor, Reduces Food Intake via Hypothalamic AMP-Activated Protein Kinase. J. Biol. Chem. 2004, 279, 19970–19976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.K.; Lin, X.; Pu, Y.; Tse, J.K.Y.; Wang, Y.; Zhang, C.L.; Cao, X.; Lau, C.W.; Huang, J.; He, L.; et al. SOX4 Is a Novel Phenotypic Regulator of Endothelial Cells in Atherosclerosis Revealed by Single-Cell Analysis. J. Adv. Res. 2022, in press. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, J.; Qu, D.; Wang, L.; Luo, J.Y.; Lau, C.W.; Liu, P.; Gao, Z.; Tipoe, G.L.; Lee, H.K.; et al. Inhibition of MiR-200c Restores Endothelial Function in Diabetic Mice Through Suppression of COX-2. Diabetes 2016, 65, 1196–1207. [Google Scholar] [CrossRef] [Green Version]
- Goh, S.Y.; Cooper, M.E. The Role of Advanced Glycation End Products in Progression and Complications of Diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products: Sparking the Development of Diabetic Vascular Injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Kassab, G.S. Assessment of Endothelial Function of Large, Medium, and Small Vessels: A Unified Myograph. Am. J. Physiol.-Heart Circ. Physiol. 2011, 300, H94–H100. [Google Scholar] [CrossRef] [Green Version]
- Gagov, H.; Gribkova, I.V.; Serebryakov, V.N.; Schubert, R. Sodium Nitroprusside-Induced Activation of Vascular Smooth Muscle BK Channels Is Mediated by PKG Rather Than by a Direct Interaction with NO. Int. J. Mol. Sci. 2022, 23, 2798. [Google Scholar] [CrossRef]
- Higashi, Y.; Maruhashi, T.; Noma, K.; Kihara, Y. Oxidative Stress and Endothelial Dysfunction: Clinical Evidence and Therapeutic Implications. Trends Cardiovasc. Med. 2014, 24, 165–169. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Tanasescu, M.; Leitzmann, M.F.; Rimm, E.B.; Hu, F.B. Physical Activity in Relation to Cardiovascular Disease and Total Mortality among Men with Type 2 Diabetes. Circulation 2003, 107, 2435–2439. [Google Scholar] [CrossRef]
- Guest, P.C.; Rahmoune, H. Characterization of the Db/Db Mouse Model of Type 2 Diabetes. Methods Mol. Biol. 2019, 1916, 195–201. [Google Scholar] [CrossRef]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-Induced Skeletal Muscle Remodeling and Metabolic Adaptation: Redox Signaling and Role of Autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef]
- Kim, B.; Lee, H.; Kawata, K.; Park, J.Y. Exercise-Mediated Wall Shear Stress Increases Mitochondrial Biogenesis in Vascular Endothelium. PLoS ONE 2014, 9, e111409. [Google Scholar] [CrossRef]
- Sun, X.; Sit, A.; Feinberg, M.W. Role for MiR-181 Family in Regulating Vascular Inflammation and Immunity. Trends Cardiovasc. Med. 2014, 24, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral Advanced Glycation Endproducts (AGEs) Promote Insulin Resistance and Diabetes by Depleting the Antioxidant Defenses AGE Receptor-1 and Sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef] [Green Version]
- Mao, W.; Fan, Y.; Wang, X.; Feng, G.; You, Y.; Li, H.; Chen, Y.; Yang, J.; Weng, H.; Shen, X. Phloretin Ameliorates Diabetes-Induced Endothelial Injury through AMPK-Dependent Anti-EndMT Pathway. Pharmacol. Res. 2022, 179, 106205. [Google Scholar] [CrossRef]
- Wu, N.; Shen, H.; Liu, H.; Wang, Y.; Bai, Y.; Han, P. Acute Blood Glucose Fluctuation Enhances Rat Aorta Endothelial Cell Apoptosis, Oxidative Stress and pro-Inflammatory Cytokine Expression in Vivo. Cardiovasc. Diabetol. 2016, 15, 109. [Google Scholar] [CrossRef] [Green Version]
- Schiffrin, E.L. Oxidative Stress, Nitric Oxide Synthase, and Superoxide Dismutase: A Matter of Imbalance Underlies Endothelial Dysfunction in the Human Coronary Circulation. Hypertension 2008, 51, 31–32. [Google Scholar] [CrossRef] [Green Version]
- Jansen, T.; Kvandová, M.; Daiber, A.; Stamm, P.; Frenis, K.; Schulz, E.; Münzel, T.; Kröller-Schön, S. The AMP-Activated Protein Kinase Plays a Role in Antioxidant Defense and Regulation of Vascular Inflammation. Antioxidants 2020, 9, 525. [Google Scholar] [CrossRef]
- Warburton, D.E.R.; Bredin, S.S.D. Health Benefits of Physical Activity: A Systematic Review of Current Systematic Reviews. Curr. Opin. Cardiol. 2017, 32, 541–556. [Google Scholar] [CrossRef]
- Joyner, M.J.; Casey, D.P. Regulation of Increased Blood Flow (Hyperemia) to Muscles During Exercise: A Hierarchy of Competing Physiological Needs. Physiol. Rev. 2015, 95, 549–601. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lee, T.S.; Kolb, E.M.; Sun, K.; Lu, X.; Sladek, F.M.; Kassab, G.S.; Garland, T.; Shyy, J.Y.J. AMP-Activated Protein Kinase Is Involved in Endothelial NO Synthase Activation in Response to Shear Stress. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1281–1287. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Kim, C.W.; Simmons, R.D.; Jo, H. Role of Flow-Sensitive MicroRNAs in Endothelial Dysfunction and Atherosclerosis Mechanosensitive Athero-MiRs. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2206–2216. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Mean ± SD |
|---|---|
| No. of patients | 12 |
| No. of diabetic/non-diabetic patients | 4/8 |
| No. of males/females | 7/5 |
| Mean age | 63.75 ± 14.25 |
| mRNA/miRNA | Forward (5′-3′) | Reverse (5′-3′) | Accession Number |
|---|---|---|---|
| ICAM1 | TTGGGCATAGAGACCCCGTT | GCACATTGCTCAGTTCATACACC | NM_000201 |
| VCAM1 | CAGTAAGGCAGGCTGTAAAAGA | TGGAGCTGGTAGACCCTCG | NM_001078 |
| IL-6 | CCTGAACCTTCCAAAGATGGC | TTCACCAGGCAAGTCTCCTCA | NM_000600 |
| GAPDH | CCACTCCTCCACCTTTGAC | ACCCTGTTGCTGTAGCCA | NM_002046 |
| mRNA/miRNA | Forward (5′-3′) | Reverse (5′-3′) | Accession Number |
|---|---|---|---|
| Icam1 | GTGATGCTCAGGTATCCATCCA | CACAGTTCTCAAAGCACAGCG | NM_010493 |
| Vcam1 | GTTCCAGCGAGGGTCTACC | AACTCTTGGCAAACATTAGGTGT | NM_011693 |
| Il-6 | TTCAGCCCTTGCTTGCCTC | ACACTTTTACTCCGAAGTCGGT | NM_031168 |
| Cdh5 | ATTGGCCTGTGTTTTCGCAC | CACAGTGGGGTCATCTGCAT | NM_009868 |
| Gapdh | AGGTCGGTGTGAACGGATTTG | TGTAGACCATGTAGTTGAGGTCA | NM_001289726 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, C.-K.; Shang, W.; Liu, J.; Cheang, W.-S.; Wang, Y.; Xiang, L.; Lau, C.-W.; Luo, J.-Y.; Ng, C.-F.; Huang, Y.; et al. Activation of AMPK/miR-181b Axis Alleviates Endothelial Dysfunction and Vascular Inflammation in Diabetic Mice. Antioxidants 2022, 11, 1137. https://doi.org/10.3390/antiox11061137
Cheng C-K, Shang W, Liu J, Cheang W-S, Wang Y, Xiang L, Lau C-W, Luo J-Y, Ng C-F, Huang Y, et al. Activation of AMPK/miR-181b Axis Alleviates Endothelial Dysfunction and Vascular Inflammation in Diabetic Mice. Antioxidants. 2022; 11(6):1137. https://doi.org/10.3390/antiox11061137
Chicago/Turabian StyleCheng, Chak-Kwong, Wenbin Shang, Jian Liu, Wai-San Cheang, Yu Wang, Li Xiang, Chi-Wai Lau, Jiang-Yun Luo, Chi-Fai Ng, Yu Huang, and et al. 2022. "Activation of AMPK/miR-181b Axis Alleviates Endothelial Dysfunction and Vascular Inflammation in Diabetic Mice" Antioxidants 11, no. 6: 1137. https://doi.org/10.3390/antiox11061137
APA StyleCheng, C.-K., Shang, W., Liu, J., Cheang, W.-S., Wang, Y., Xiang, L., Lau, C.-W., Luo, J.-Y., Ng, C.-F., Huang, Y., & Wang, L. (2022). Activation of AMPK/miR-181b Axis Alleviates Endothelial Dysfunction and Vascular Inflammation in Diabetic Mice. Antioxidants, 11(6), 1137. https://doi.org/10.3390/antiox11061137