



Optimization of Ursolic Acid Extraction in Oil from Annurca Apple to Obtain Oleolytes with Potential Cosmeceutical Application

 ,
,  , ,
, ,  ,
,  , , and
, , and 
Abstract
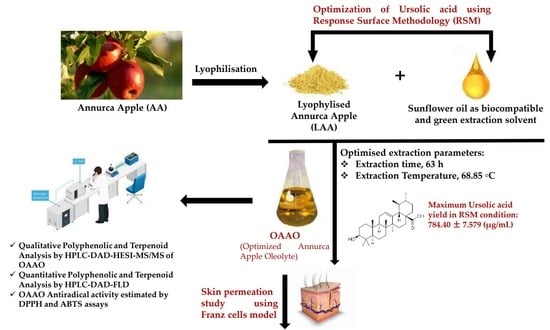
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Sample Collection and Oleolyte Preparation
2.2.1. Oleolyte Preparation Protocol and Experimental Design
2.2.2. Oil Deacidification Procedure
2.2.3. Acidity Determination of Oil Samples
2.3. Triterpenoic Analysis
2.3.1. Ursolic Acid Extraction Protocol
2.3.2. Ursolic Acid Quantitative Analysis by HPLC-DAD
2.3.3. Linearity and Sensitivity of the Ursolic Acid HPLC-DAD Analysis
2.3.4. Accuracy and Precision of Ursolic Acid HPLC-DAD Analysis
2.3.5. Matrix Effect of Ursolic Acid Extraction
2.3.6. Recovery of Ursolic Acid Extraction
2.4. Polyphenols Analysis
2.4.1. Polyphenolic Extraction
2.4.2. Polyphenolic Quantitative Analysis by HPLC-DAD Analysis
2.5. OAAO Qualitative Composition by HPLC-DAD-HESI-MS/MS Analysis
2.6. Total Phenolic Content Determination
2.7. Antioxidant Activity
2.7.1. DPPH• Radical Scavenging Assay
2.7.2. Ferric Reducing/Antioxidant Power (FRAP) Assay
2.7.3. ABTS• Radical Scavenging Assay
2.8. Skin Sample
2.8.1. Diffusion Experiment
2.8.2. HPLC-MS Method for Franz’s Diffusion Cell System
2.9. Statistics
3. Results and Discussion
3.1. Optimisation of Ursolic Acid Extraction Using RSM Model
3.2. Quantitative Polyphenolic Analysis of OAAO by HPLC-DAD-FLD
3.3. Qualitative Polyphenols and Terpenoid Characterisation by HPLC-HESI-MS/MS
3.4. Validation of Ursolic Acid HPLC-DAD Analysis Method
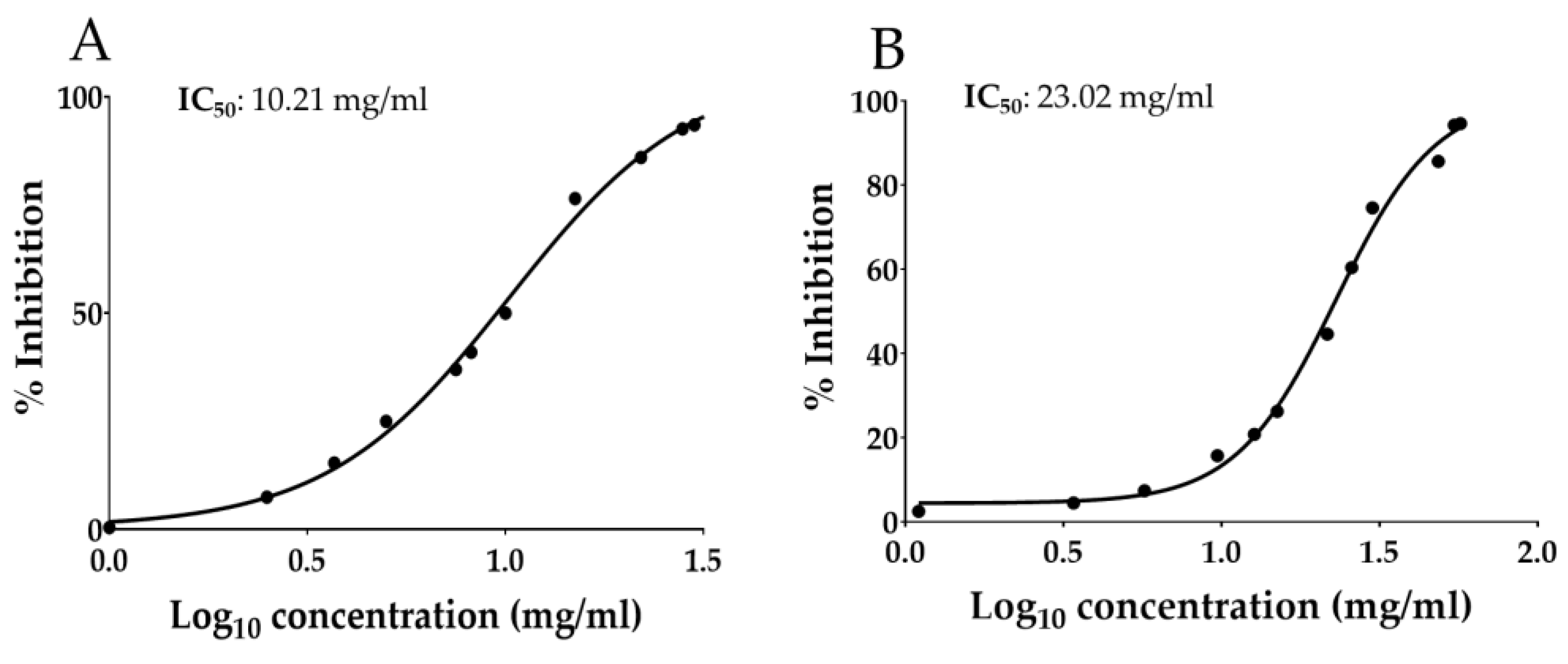
3.5. Total Polyphenols and In Vitro Antiradical Activity of OAAO
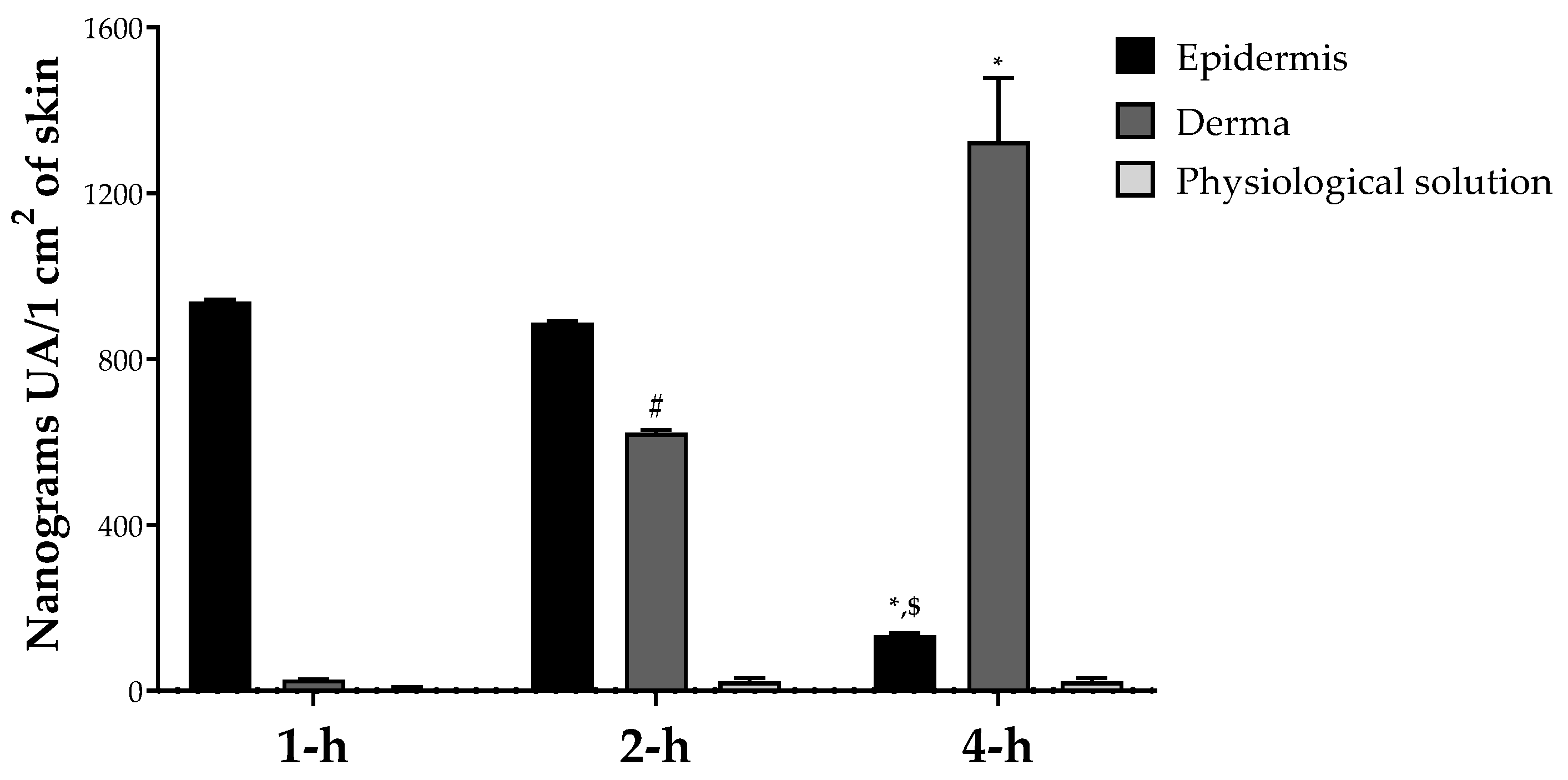
3.6. Skin Permeation Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- López-Hortas, L.; Pérez-Larrán, P.; González-Muñoz, M.J.; Falqué, E.; Domínguez, H. Recent Developments on the Extraction and Application of Ursolic Acid. A Review. Food Res. Int. 2018, 103, 130–149. [Google Scholar] [CrossRef] [PubMed]
- Cargnin, S.T.; Gnoatto, S.B. Ursolic Acid from Apple Pomace and Traditional Plants: A Valuable Triterpenoid with Functional Properties. Food Chem 2017, 220, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Dini, I.; Falanga, D.; di Lorenzo, R.; Tito, A.; Carotenuto, G.; Zappelli, C.; Grumetto, L.; Sacchi, A.; Laneri, S.; Apone, F. An Extract from Ficus Carica Cell Cultures Works as an Anti-Stress Ingredient for the Skin. Antioxidants 2021, 10, 1–21. [Google Scholar] [CrossRef]
- Laneri, S.; Dini, I.; Tito, A.; di Lorenzo, R.; Bimonte, M.; Tortora, A.; Zappelli, C.; Angelillo, M.; Bernardi, A.; Sacchi, A.; et al. Plant Cell Culture Extract of Cirsium Eriophorum with Skin Pore Refiner Activity by Modulating Sebum Production and Inflammatory Response. Phytother. Res. 2021, 35, 530–540. [Google Scholar] [CrossRef]
- Laneri, S.; di Lorenzo, R.; Sacchi, A.; Dini, I. Dosage of Bioactive Molecules in the Nutricosmeceutical Helix Aspersa Muller Mucus and Formulation of New Cosmetic Cream with Moisturizing Effect. Nat. Prod. Commun. 2019, 14, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Laneri, S.; di Lorenzo, R.M.; Bernardi, A.; Sacchi, A.; Dini, I. Aloe Barbadensis: A Plant of Nutricosmetic Interest. Nat. Prod. Commun. 2020, 15. [Google Scholar] [CrossRef]
- Both, D.M.; Goodtzova, K.; Yarosh, D.B.; Brown, D.A. Liposome-Encapsulated Ursolic Acid Increases Ceramides and Collagen in Human Skin Cells. Arch. Dermatol. Res. 2002, 293, 569–575. [Google Scholar] [CrossRef]
- Wójciak-Kosior, M.; Paduch, R.; Matysik-Woźniak, A.; Niedziela, P.; Donica, H. The Effect of Ursolic and Oleanolic Acids on Human Skin Fibroblast Cells. Folia Histochem. Cytobiol. 2011, 49, 664–669. [Google Scholar] [CrossRef] [Green Version]
- Yo, K.; Oba, A.; Tada, A. Novel Approach for Improving Skin Roughness Mediated by Ker- Atin Intermediate Filaments. J. Dermatol. Sci. 2013, 69, e51. [Google Scholar] [CrossRef]
- Soleymani, S.; Farzaei, M.H.; Zargaran, A.; Niknam, S.; Rahimi, R. Promising Plant-Derived Secondary Metabolites for Treatment of Acne Vulgaris: A Mechanistic Review. Arch. Dermatol. Res. 2020, 312, 5–23. [Google Scholar] [CrossRef]
- Neimkhum, W.; Anuchapreeda, S.; Lin, W.C.; Lue, S.C.; Lee, K.H.; Chaiyana, W. Effects of Carissa Carandas Linn. Fruit, Pulp, Leaf, and Seed on Oxidation, Inflammation, Tyrosinase, Matrix Metalloproteinase, Elastase, and Hyaluronidase Inhibition. Antioxidants 2021, 10, 1345. [Google Scholar] [CrossRef] [PubMed]
- Guinda, A.; Rada, M.; Delgado, T.; Castellano, J.M. Pentacyclic Triterpenic Acids from Argania Spinosa. Eur. J. Lipid Sci. Technol. 2011, 113, 231–237. [Google Scholar] [CrossRef]
- Cui, T.; Li, J.Z.; Kayahara, H.; Ma, L.; Wu, L.X.; Nakamura, K. Quantification of the Polyphenols and Triterpene Acids in Chinese Hawthorn Fruit by High-Performance Liquid Chromatography. J. Agric. Food Chem. 2006, 54, 4574–4581. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Mackinnon, S.L.; Craft, C.C.; Matchett, M.D.; Hurta, R.A.R.; Neto, C.C. Ursolic Acid and Its Esters: Occurrence in Cranberries and Other Vaccinium Fruit and Effects on Matrix Metalloproteinase Activity in DU145 Prostate Tumor Cells. J. Sci. Food Agric. 2011, 91, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Duan, J.A.; Qian, D.; Tang, Y.; Wu, D.; Su, S.; Wang, H.; Zhao, Y. Content Variations of Triterpenic Acid, Nucleoside, Nucleobase, and Sugar in Jujube (Ziziphus Jujuba) Fruit during Ripening. Food Chem. 2015, 167, 468–474. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Noshita, T.; Kidachi, Y.; Umetsu, H.; Hayashi, M.; Komiyama, K.; Funayama, S.; Ryoyama, K. Isolation of Ursolic Acid from Apple Peels and Its Specific Efficacy as a Potent Antitumor Agent. J. Health Sci. 2008, 54, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Tenore, G.C.; Campiglia, P.; Stiuso, P.; Ritieni, A.; Novellino, E. Nutraceutical Potential of Polyphenolic Fractions from Annurca Apple (M. Pumila Miller Cv Annurca). Food Chem. 2013, 140, 614–622. [Google Scholar] [CrossRef]
- Tenore, G.C.; Calabrese, G.; Stiuso, P.; Ritieni, A.; Giannetti, D.; Novellino, E. Effects of Annurca Apple Polyphenols on Lipid Metabolism in HepG2 Cell Lines: A Source of Nutraceuticals Potentially Indicated for the Metabolic Syndrome. Food Res. Int. 2014, 63, 252–257. [Google Scholar] [CrossRef]
- Tenore, G.C.; Caruso, D.; Buonomo, G.; D’Avino, M.; Campiglia, P.; Marinelli, L.; Novellino, E. A Healthy Balance of Plasma Cholesterol by a Novel Annurca Apple-Based Nutraceutical Formulation: Results of a Randomized Trial. J. Med. Food. 2017, 20, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Riccio, G.; Maisto, M.; Bottone, S.; Badolati, N.; Rossi, G.B.; Tenore, G.C.; Stornaiuolo, M.; Novellino, E. WNT Inhibitory Activity of Malus Pumila Miller Cv Annurca and Malus Domestica Cv Limoncella Apple Extracts on Human Colon-Rectal Cells Carrying Familial Adenomatous Polyposis Mutations. Nutrients 2017, 9, 1262. [Google Scholar] [CrossRef]
- D’Abrosca, B.; Fiorentino, A.; Monaco, P.; Oriano, P.; Pacifico, S. Annurcoic Acid: A New Antioxidant Ursane Triterpene from Fruits of Cv. Annurca Apple. Food Chem. 2006, 98, 285–290. [Google Scholar] [CrossRef]
- da Silva, G.N.S.; Maria, N.R.G.; Schuck, D.C.; Cruz, L.N.; de Moraes, M.S.; Nakabashi, M.; Graebin, C.; Gosmann, G.; Garcia, C.R.S.; Gnoatto, S.C.B. Two Series of New Semisynthetic Triterpene Derivatives: Differences in Anti-Malarial Activity, Cytotoxicity and Mechanism of Action. Malar. J. 2013, 12, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innocente, A.M.; Silva, G.N.S.; Cruz, L.N.; Moraes, M.S.; Nakabashi, M.; Sonnet, P.; Gosmann, G.; Garcia, C.R.S.; Gnoatto, S.C.B. Synthesis and Antiplasmodial Activity of Betulinic Acid and Ursolic Acid Analogues. Molecules 2012, 17, 12003–12014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- lo Scalzo, R.; Testoni, A.; Genna, A. “Annurca” Apple Fruit, a Southern Italy Apple Cultivar: Textural Properties and Aroma Composition. Food Chem. 2001, 73, 333–343. [Google Scholar] [CrossRef]
- Prasanth Kumar, P.K.; Gopala Krishna, A.G. Impact of Different Deacidification Methods on Quality Characteristics and Composition of Olein and Stearin in Crude Red Palm Oil. J. Oleo Sci. 2014, 63, 1209–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontogianni, V.G.; Tomic, G.; Nikolic, I.; Nerantzaki, A.A.; Sayyad, N.; Stosic-Grujicic, S.; Stojanovic, I.; Gerothanassis, I.P.; Tzakos, A.G. Phytochemical Profile of Rosmarinus Officinalis and Salvia Officinalis Extracts and Correlation to Their Antioxidant and Anti-Proliferative Activity. Food Chem. 2013, 136, 120–129. [Google Scholar] [CrossRef]
- Maisto, M.; Schiano, E.; Novellino, E.; Piccolo, V.; Iannuzzo, F.; Salviati, E.; Summa, V.; Annunziata, G.; Tenore, G.C. Application of a Rapid and Simple Technological Process to Increase Levels and Bioccessibility of Free Phenolic Compounds in Annurca Apple Nutraceutical Product. Foods 2022, 11, 1453. [Google Scholar] [CrossRef]
- Ohno, Y. ICH Guidelines—Implementation of the 3Rs (Refinement, Reduction, and Replacement): Incorporating Best Scientific Practices into the Regulatory Process. ILAR J. 2002, 43, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Assiri, A.M.A.; Elbanna, K.; Abulreesh, H.H.; Ramadan, M.F. Bioactive Compounds of Cold-Pressed Thyme (Thymus Vulgaris) Oil with Antioxidant and Antimicrobial Properties. J. Oleo Sci. 2016, 65, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Maisto, M.; Iannuzzo, F.; Schiano, E.; Ciampaglia, R.; Labanca, A.; Montesano, D.; Piccolo, V.; Rossi, P.; Tenore, G.C. Effects of Fortified Laying Hen Diet with Moringa Oleifera Leaves and Goji Berries on Cholesterol and Carotenoid Egg Content. Foods 2022, 11, 3156. [Google Scholar] [CrossRef]
- Maisto, M.; Annunziata, G.; Schiano, E.; Piccolo, V.; Iannuzzo, F.; Santangelo, R.; Ciampaglia, R.; Tenore, G.C.; Novellino, E.; Grieco, P. Potential Functional Snacks: Date Fruit Bars Supplemented by Different Species of Lactobacillus Spp. Foods 2021, 10, 1760. [Google Scholar] [CrossRef]
- Neri, I.; Laneri, S.; di Lorenzo, R.; Dini, I.; Russo, G.; Grumetto, L. Parabens Permeation through Biological Membranes: A Comparative Study Using Franz Cell Diffusion System and Biomimetic Liquid Chromatography. Molecules 2022, 27, 4263. [Google Scholar] [CrossRef]
- di Lorenzo, R.; Bernardi, A.; Grumetto, L.; Sacchi, A.; Avagliano, C.; Coppola, S.; Severina, A.F. de G. di S.; Bruno, C.; Paparo, L.; Laneri, S.; et al. Phenylalanine Butyramide Is a New Cosmetic Ingredient with Soothing and Anti-Reddening Potential. Molecules 2021, 26, 6611. [Google Scholar] [CrossRef] [PubMed]
- Xia, E.Q.; Yu, Y.Y.; Xu, X.R.; Deng, G.F.; Guo, Y.J.; Li, H. Ultrasound-Assisted Extraction of Oleanolic Acid and Ursolic Acid from Ligustrum Lucidum Ait. Ultrason. Sonochem. 2012, 19, 772–776. [Google Scholar] [CrossRef]
- Li, G.; Zhang, X.; You, J.; Song, C.; Sun, Z.; Xia, L.; Suo, Y. Highly Sensitive and Selective Pre-Column Derivatization High-Performance Liquid Chromatography Approach for Rapid Determination of Triterpenes Oleanolic and Ursolic Acids and Application to Swertia Species: Optimization of Triterpenic Acids Extraction and Pre-Column Derivatization Using Response Surface Methodology. Anal. Chim. Acta 2011, 688, 208–218. [Google Scholar] [CrossRef]
- Fu, Q.; Zhang, L.; Cheng, N.; Jia, M.; Zhang, Y. Extraction Optimization of Oleanolic and Ursolic Acids from Pomegranate (Punica Granatum L.) Flowers. Food Bioprod. Process. 2014, 92, 321–327. [Google Scholar] [CrossRef]
- Vetal, M.D.; Lade, V.G.; Rathod, V.K. Extraction of Ursolic Acid from Ocimum Sanctum by Ultrasound: Process Intensification and Kinetic Studies. Chem. Eng. Process. Process Intensif. 2013, 69, 24–30. [Google Scholar] [CrossRef]
- Miron, T.L.; Plaza, M.; Bahrim, G.; Ibáñez, E.; Herrero, M. Chemical Composition of Bioactive Pressurized Extracts of Romanian Aromatic Plants. J. Chromatogr. A 2011, 1218, 4918–4927. [Google Scholar] [CrossRef] [Green Version]
- Vergara-Salinas, J.R.; Pérez-Jiménez, J.; Torres, J.L.; Agosin, E.; Pérez-Correa, J.R. Effects of Temperature and Time on Polyphenolic Content and Antioxidant Activity in the Pressurized Hot Water Extraction of Deodorized Thyme (Thymus Vulgaris). J. Agric. Food Chem. 2012, 60, 10920–10929. [Google Scholar] [CrossRef]
- Maisto, M.; Piccolo, V.; Novellino, E.; Schiano, E.; Iannuzzo, F.; Ciampaglia, R.; Summa, V.; Tenore, G.C. Optimization of Phlorizin Extraction from Annurca Apple Tree Leaves Using Response Surface Methodology. Antioxidants 2022, 11, 1933. [Google Scholar] [CrossRef]
- Li, Z.H.; Guo, H.; Xu, W.-B.; Ge, J.; Li, X.; Alimu, M.; He, D.J. Rapid Identification of Flavonoid Constituents Directly from PTP1B Inhibitive Extract of Raspberry (Rubus Idaeus L.) Leaves by HPLC-ESI-QTOF-MS-MS. J. Chromatogr. Sci. 2016, 54, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Singh, A.; Kumar, B. Identification and Characterization of Phenolics and Terpenoids from Ethanolic Extracts of Phyllanthus Species by HPLC-ESI-QTOF-MS/MS. J. Pharm. Anal. 2017, 7, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Abdelaziz, S.; Hassan, W.H.B.; Elhassanny, A.E.M.; Al-Yousef, H.M.; Elsayed, M.A.; Adel, R. Ultra Performance Liquid Chromatography-Tandem Mass Spectrometeric Analysis of Ethyl Acetate Fraction from Saudi Lavandula Coronopifolia Poir and Evaluation of Its Cytotoxic and Antioxidant Activities. J. HerbMed Pharmacol. 2020, 9, 268–276. [Google Scholar] [CrossRef]
- Huhman, D.v.; Berhow, M.A.; Sumner, L.W. Quantification of Saponins in Aerial and Subterranean Tissues of Medicago Truncatula. J. Agric. Food Chem. 2005, 53, 1914–1920. [Google Scholar] [CrossRef]
- Zou, D.; Wang, J.; Zhang, B.; Xie, S.; Wang, Q.; Xu, K.; Lin, R.; Lam, C.W.K. Analysis of Chemical Constituents in Wuzi-Yanzong-Wan by UPLC-ESI-LTQ-Orbitrap-MS. Molecules 2015, 20, 21373–21404. [Google Scholar] [CrossRef]
- Guo, Y.P.; Yang, H.; Wang, Y.L.; Chen, X.X.; Zhang, K.; Wang, Y.L.; Sun, Y.F.; Huang, J.; Yang, L.; Wang, J.H. Determination of Flavonoids Compounds of Three Species and Different Harvesting Periods in Crataegi Folium Based on Lc-Ms/Ms. Molecules 2021, 26, 1602. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liao, Y.; Fang, C.; Tsunoda, M.; Zhang, Y.; Song, Y.; Deng, S. Simultaneous Analysis of Ursolic Acid and Oleanolic Acid in Guava Leaves Using QuEChERS-Based Extraction Followed by High-Performance Liquid Chromatography. J. Anal. Methods Chem. 2017, 2017, 2984562. [Google Scholar] [CrossRef] [Green Version]
- Zillich, O.v.; Schweiggert-Weisz, U.; Eisner, P.; Kerscher, M. Polyphenols as Active Ingredients for Cosmetic Products. Int. J. Cosmet. Sci. 2015, 37, 455–464. [Google Scholar] [CrossRef]
- Luo, J.; Si, H.; Jia, Z.; Liu, D. Dietary Anti-Aging Polyphenols and Potential Mechanisms. Antioxidants 2021, 10, 1–20. [Google Scholar] [CrossRef]
- Graziani, G.; Gaspari, A.; di Vaio, C.; Cirillo, A.; Ronca, C.L.; Grosso, M.; Ritieni, A. Assessment of In Vitro Bioaccessibility of Polyphenols from Annurca, Limoncella, Red Delicious, and Golden Delicious Apples Using a Sequential Enzymatic Digestion Model. Antioxidants 2021, 10, 541. [Google Scholar] [CrossRef]
- Schiano, E.; Piccolo, V.; Novellino, E.; Maisto, M.; Iannuzzo, F.; Summa, V.; Tenore, G.C. Thinned Nectarines, an Agro-Food Waste with Antidiabetic Potential: HPLC-HESI-MS/MS Phenolic Characterization and In Vitro Evaluation of Their Beneficial Activities. Foods 2022, 11, 1010. [Google Scholar] [CrossRef] [PubMed]
- Vega, E.; Egea, M.A.; Garduño-Ramírez, M.L.; García, M.L.; Sánchez, E.; Espina, M.; Calpena, A.C. Flurbiprofen PLGA-PEG Nanospheres: Role of Hydroxy-β-Cyclodextrin on Ex Vivo Human Skin Permeation and in Vivo Topical Anti-Inflammatory Efficacy. Colloids Surf B Biointerfaces 2013, 110, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic-curic, N.; Maibach, H.I. Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancemen; Springer: New York, NY, USA, 2015; ISBN 9783662450123. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Factor Levels | ||||||
|---|---|---|---|---|---|---|---|
| Time (hours) | 1 | 2 | 4 | 12 | 24 | 48 | 96 |
| Temperature (°C) | 20 | 40 | 60 | 80 | |||
| Total runs | 28 | ||||||
| Compound | Mean Value ± SD (µg/mL of OAAO) |
|---|---|
| Chlorogenic acid | nd |
| Caffeic acid | nd |
| p-Cumaric acid | nd |
| Procyanidin B1 + B3 | nd |
| Procyanidin B2 | nd |
| Epicatechin | nd |
| Rutin | 0.76 ± 0.001 |
| Quercetin-3-O-glucoside | 0.71 ± 0.007 |
| Kaempferol-3-O-rhamnoside | 0.15 ± 0.010 |
| Kaempferol-3-O-glucoside | LOQ |
| Apigenin-7-O-glucoside | 0.0081 ± 0.0001 |
| Phloridzin | 0.15 ± 0.010 |
| Quercetin | nd |
| Phloretin | 0.07 ± 0.01 |
| No. | Extract | Compound | Ione | Rt | m/z | Diagnostic Fragment | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Hydroalcoholic | Rutin | [M-H]− | 8.05 | 609 | 591 [M-H-H2O]−, 301 [M-H-Rut]−, 255 [M-H-Rut-CO-H2O]−, 179 [M-H-Rut-C7H6O2]− | [41] |
| 2 | Hydroalcoholic | Quercetin-3-O-glucoside | [M-H]− | 8.16 | 463 | 445 [M-H-H2O]−, 301 [M-H-Glu]−, 255 [M-H-Glu-CO-H2O]−, 179 [M-H-Glu-C7H6O2]− | [4] |
| 3 | Hydroalcoholic | Kaempferol-3-O-glucoside | [M-H]− | 8.73 | 447 | 429 [M-H-H2O]−, 285 [M-H-Glu]−, 179 [M-H-Glu-C7H6O2]−, 151 [M-H-Glu-C8H6O3]− | [41] |
| 4 | Hydroalcoholic | Phloridzin | [M-H]− | 9.41 | 435 | 417 [M-H-H2O]−, 273 [M-H-Glu]−, 167 [M-H-Glu-C13H16O6]− | [40] |
| 5 | Hydroalcoholic | Kaempferol-3-O-rhamnoside | [M-H]− | 9.78 | 431 | 413 [M-H-H2O]−, 327 [M-H-C4H8O3]−, 285 [M-H-Rha]−, 179 [M-H-Rha-C7H6O2]− | [42] |
| 6 | Ethylacetate | Hydroxymethoxyursolic acid | [M+H]+ | 11.49 | 503 | 485 [M+H-H2O]+, 467 [M+H-2H2O]−, 457 [M+H-HCO2H]+, 439 [M+H-HCO2H-H2O]+ | [43] |
| 7 | Hydroalcoholic | Phloretin | [M-H]− | 11.61 | 273 | 255 [M-H-H2O]−, 167 [M-H-C7H6O]−, 125 [M-H-C9H8O2]− | [40] |
| 8 | Ethylacetate | Carboxyursolic acid | [M+H]+ | 12.17 | 501 | 483 [M+H-H2O]+, 455 [M+H-HCO2H]+, 437 [M+H-HCO2H-H2O]+, 419 [M+H-HCO2H-2H2O]+ | [43] |
| 9 | Ethylacetate | Annurcoic acid | [M+H]+ | 13.47 | 487 | 469 [M+H-H2O]+, 451 [M+H-2H2O]−, 441 [M+H-HCO2H]+, 423 [M+H-HCO2H-H2O]+ | [20] |
| 10 | Ethylacetate | Zanhic acid | [M-H]− | 14.83 | 517 | 499 [M-H-H2O]−, 473 [M-H-CO2]−, 455 [M-H-CO2-H2O]−, 429 [M-H-2CO2]− | [44] |
| 11 | Ethylacetate | Medicagenic acid isomer 1 | [M-H]− | 15.09 | 501 | 483 [M-H-H2O]−, 457 [M-H-CO2]−, 409 [M-H-2HCOOH]−, 391 [M-H-2HCOOH-H2O]− | [44] |
| 12 | Ethylacetate | Corosolic acid | [M+H]+ | 15.19 | 473 | 455 [M+H-H2O]+, 427 [M+H-HCO2H]+, 409 [M+H-HCO2H-H2O]+, 391 [M+H-HCO2H-2H2O]+ | [45] |
| 13 | Ethylacetate | Arjunolic acid | [M-H]− | 15.75 | 487 | 469 [M-H-H2O]−, 441 [M-H-HCOOH]−, 425 [M-H-CO2-H2O]−, 407 [M-H-CO2-2H2O]− | [45] |
| 14 | Ethylacetate | Oleanolic acid | [M+H]+ | 15.91 | 457 | 439 [M+H-H2O]+, 411 [M+H-HCO2H]+, 393 [M+H-HCO2H-H2O]+ | [45] |
| 15 | Ethylacetate | Ursolic acid | [M+H]+ | 16.20 | 457 | 439 [M+H-H2O]+, 411 [M+H-HCO2H]+, 393 [M+H-HCO2H-H2O]+ | [45] |
| 16 | Ethylacetate | Medicagenic acid isomer 2 | [M-H]− | 16.67 | 501 | 483 [M-H-H2O]−, 457 [M-H-CO2]−, 439 [M-H-CO2-H2O]−, 391 [M-H-2HCOOH-H2O]− | [44] |
| 17 | Ethylacetate | Dihydroxy-{[(hydroxyphenyl)-propenoyl)]oxy}ursenoic acid | [M-H]− | 18.79 | 633 | 615 [M-H-H2O]−, 589 [M-H-CO2]−, 571 [M-H-CO2-H2O]−, 487 [M-H-CA]− | [46] |
| 18 | Ethylacetate | Hydroxy-{[(hydroxyphenyl)-propenoyl)]oxy}ursenoic acid | [M-H]− | 20.89 | 617 | 599 [M-H-H2O]−, 573 [M-H-CO2]−, 453 [M-H-CA-H2O]− | [46] |
| Compound | Linearity | Correlation Coefficient (r2) | LOQ (ppm) | LOD (ppm) | Monitoring Channel |
|---|---|---|---|---|---|
| Ursolic acid | Y = 4 E + 06 x + 32,787 | 0.999 | 0.845 | 0.295 | 205 nm |
| Analyte Concentration (ppm) | Intra-Day (CV%, n = 3) | Inter-Day (CV%, n = 3) | Intra-Day (%Bias, n = 3) | Inter-Day (%Bias, n = 3) | |
|---|---|---|---|---|---|
| 500 | 1.874 | 1.720 | 3.142 | 1.386 | |
| Ursolic acid | 100 | 1.960 | 0.885 | 0.580 | 1.120 |
| 5 | 5.773 | 7.375 | −0.534 | −0.521 | |
| Analyte Ursolic Acid Spiked (µg) | Recovery (%) | Matrix Effect (%) | |
|---|---|---|---|
| 30 | 89.65 | 2.29 | |
| Ursolic acid | 20 | 89.42 | 1.42 |
| 10 | 91.21 | 6.86 | |
| Antioxidant Activity (µmol TE/g of OAAO Hydroalcoholic Extract ± SD) | ||
|---|---|---|
| DPPH Assay | ABTS Assay | FRAP Assay |
| 14.63 ± 0.22 | 5.90 ± 0.49 | 21.72 ± 0.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maisto, M.; Piccolo, V.; Novellino, E.; Schiano, E.; Iannuzzo, F.; Ciampaglia, R.; Summa, V.; Tenore, G.C. Optimization of Ursolic Acid Extraction in Oil from Annurca Apple to Obtain Oleolytes with Potential Cosmeceutical Application. Antioxidants 2023, 12, 224. https://doi.org/10.3390/antiox12020224
Maisto M, Piccolo V, Novellino E, Schiano E, Iannuzzo F, Ciampaglia R, Summa V, Tenore GC. Optimization of Ursolic Acid Extraction in Oil from Annurca Apple to Obtain Oleolytes with Potential Cosmeceutical Application. Antioxidants. 2023; 12(2):224. https://doi.org/10.3390/antiox12020224
Chicago/Turabian StyleMaisto, Maria, Vincenzo Piccolo, Ettore Novellino, Elisabetta Schiano, Fortuna Iannuzzo, Roberto Ciampaglia, Vincenzo Summa, and Gian Carlo Tenore. 2023. "Optimization of Ursolic Acid Extraction in Oil from Annurca Apple to Obtain Oleolytes with Potential Cosmeceutical Application" Antioxidants 12, no. 2: 224. https://doi.org/10.3390/antiox12020224
APA StyleMaisto, M., Piccolo, V., Novellino, E., Schiano, E., Iannuzzo, F., Ciampaglia, R., Summa, V., & Tenore, G. C. (2023). Optimization of Ursolic Acid Extraction in Oil from Annurca Apple to Obtain Oleolytes with Potential Cosmeceutical Application. Antioxidants, 12(2), 224. https://doi.org/10.3390/antiox12020224