
CaMKII-Dependent Contractile Dysfunction and Pro-Arrhythmic Activity in a Mouse Model of Obstructive Sleep Apnea

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. OSA-Induction by PTFE Injection
2.2. Transthoracic Echocardiography
2.3. Isolation of Ventricular Cardiomyocytes
2.4. Measurements of Reactive Oxygen Species (ROS)
2.5. Epifluorescence Microscopy
2.6. Statistical Analysis
3. Results
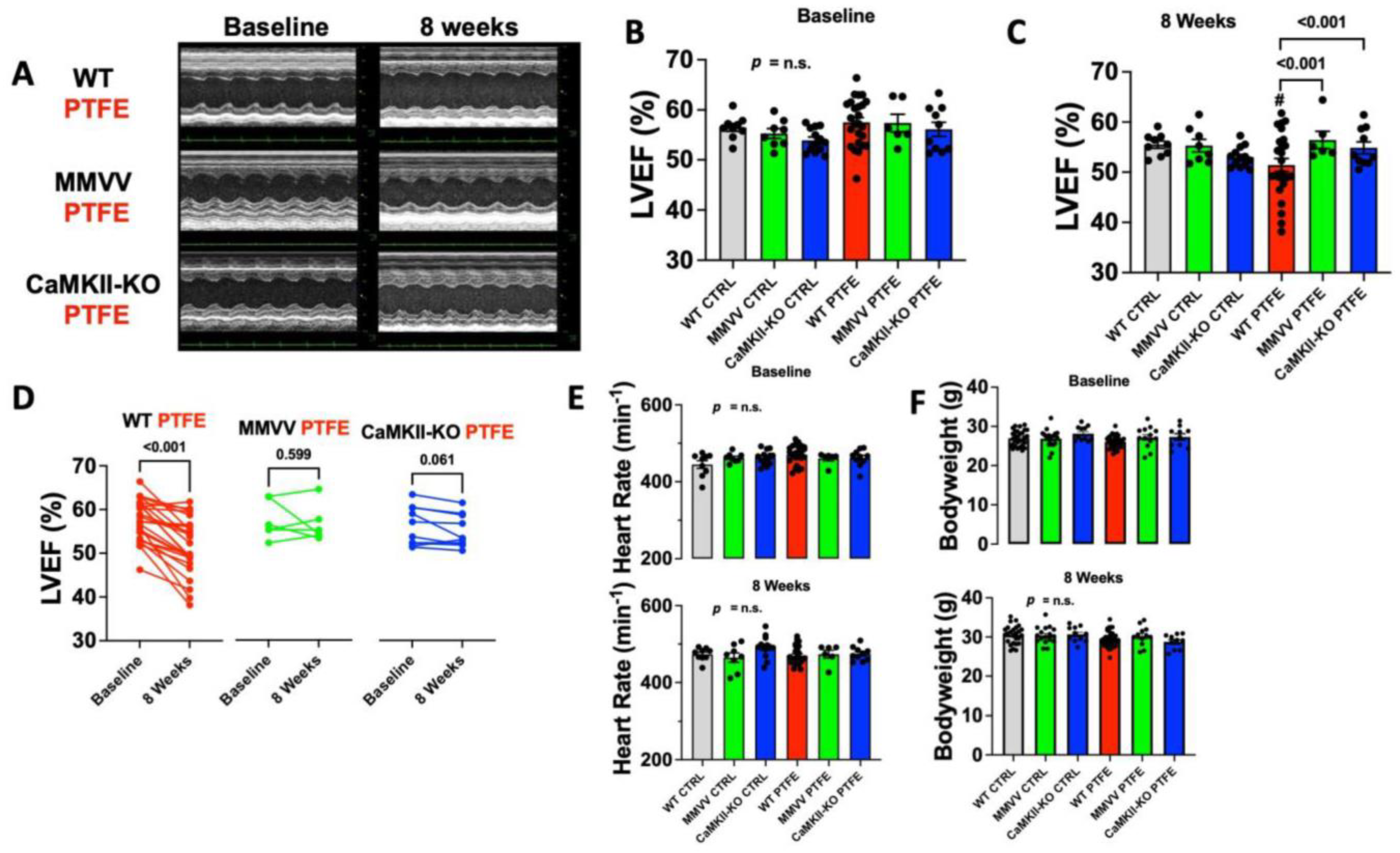
3.1. CaMKII-Dependent Contractile Dysfunction in OSA Mice
3.2. ROS Production Is Increased after PTFE Treatment
3.3. CaMKII-Dependent Dysregulation of Cellular Ca Homeostasis in OSA Mice
4. Discussion
4.1. Mechanisms of Cardiac Disease in SDB
4.2. CaMKII-Dependent Dysregulation of Cellular Ca Homeostasis
4.3. CaMKII Inhibition as a Potential Therapeutic Strategy in SDB
4.4. Study Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip, M.S.M.; Morrell, M.J.; Nunez, C.M.; Patel, S.R.; Penzel, T.; Pepin, J.L.; et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: A literature-based analysis. Lancet Respir. Med. 2019, 7, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Pengo, M.F.; Soranna, D.; Giontella, A.; Perger, E.; Mattaliano, P.; Schwarz, E.I.; Lombardi, C.; Bilo, G.; Zambon, A.; Steier, J.; et al. Obstructive sleep apnoea treatment and blood pressure: Which phenotypes predict a response? A systematic review and meta-analysis. Eur. Respir. J. 2020, 55, 1901945. [Google Scholar] [CrossRef]
- Gami, A.S.; Hodge, D.O.; Herges, R.M.; Olson, E.J.; Nykodym, J.; Kara, T.; Somers, V.K. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J. Am. Coll. Cardiol. 2007, 49, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Mehra, R.; Chung, M.K.; Olshansky, B.; Dobrev, D.; Jackson, C.L.; Kundel, V.; Linz, D.; Redeker, N.S.; Redline, S.; Sanders, P.; et al. Sleep-Disordered Breathing and Cardiac Arrhythmias in Adults: Mechanistic Insights and Clinical Implications: A Scientific Statement from the American Heart Association. Circulation 2022, 146, e119–e136. [Google Scholar] [CrossRef]
- Arzt, M.; Young, T.; Finn, L.; Skatrud, J.B.; Bradley, T.D. Association of sleep-disordered breathing and the occurrence of stroke. Am. J. Respir. Crit. Care Med. 2005, 172, 1447–1451. [Google Scholar] [CrossRef] [Green Version]
- Lebek, S.; Hegner, P.; Tafelmeier, M.; Rupprecht, L.; Schmid, C.; Maier, L.S.; Arzt, M.; Wagner, S. Female Patients with Sleep-Disordered Breathing Display More Frequently Heart Failure With Preserved Ejection Fraction. Front Med. 2021, 8, 675987. [Google Scholar] [CrossRef]
- Arzt, M.; Woehrle, H.; Oldenburg, O.; Graml, A.; Suling, A.; Erdmann, E.; Teschler, H.; Wegscheider, K.; Schla, H.F.I. Prevalence and Predictors of Sleep-Disordered Breathing in Patients with Stable Chronic Heart Failure: The SchlaHF Registry. JACC Heart Fail. 2016, 4, 116–125. [Google Scholar] [CrossRef]
- Arzt, M.; Oldenburg, O.; Graml, A.; Schnepf, J.; Erdmann, E.; Teschler, H.; Schoebel, C.; Woehrle, H.; SchlaHF-XT Investigators. Prevalence and predictors of sleep-disordered breathing in chronic heart failure: The SchlaHF-XT registry. ESC Heart Fail. 2022, 9, 4100–4111. [Google Scholar] [CrossRef]
- Peker, Y.; Glantz, H.; Eulenburg, C.; Wegscheider, K.; Herlitz, J.; Thunstrom, E. Effect of Positive Airway Pressure on Cardiovascular Outcomes in Coronary Artery Disease Patients with Nonsleepy Obstructive Sleep Apnea. The RICCADSA Randomized Controlled Trial. Am. J. Respir. Crit. Care Med. 2016, 194, 613–620. [Google Scholar] [CrossRef]
- Zinchuk, A.V.; Chu, J.H.; Liang, J.; Celik, Y.; Op de Beeck, S.; Redeker, N.S.; Wellman, A.; Yaggi, H.K.; Peker, Y.; Sands, S.A. Physiological Traits and Adherence to Sleep Apnea Therapy in Individuals with Coronary Artery Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 703–712. [Google Scholar] [CrossRef]
- Lebek, S.; Pichler, K.; Reuthner, K.; Trum, M.; Tafelmeier, M.; Mustroph, J.; Camboni, D.; Rupprecht, L.; Schmid, C.; Maier, L.S.; et al. Enhanced CaMKII-Dependent Late INa Induces Atrial Proarrhythmic Activity in Patients with Sleep-Disordered Breathing. Circ. Res. 2020, 126, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Knezl, V.; Okruhlicova, L.; Slezak, J. Myocardial gap junctions: Targets for novel approaches in the prevention of life-threatening cardiac arrhythmias. Physiol. Res. 2008, 57, S1–S13. [Google Scholar] [CrossRef] [PubMed]
- Andelova, K.; Egan Benova, T.; Szeiffova Bacova, B.; Sykora, M.; Prado, N.J.; Diez, E.R.; Hlivak, P.; Tribulova, N. Cardiac Connexin-43 Hemichannels and Pannexin1 Channels: Provocative Antiarrhythmic Targets. Int. J. Mol. Sci. 2020, 22, 260. [Google Scholar] [CrossRef] [PubMed]
- Andelova, K.; Szeiffova Bacova, B.; Sykora, M.; Pavelka, S.; Rauchova, H.; Tribulova, N. Cardiac Cx43 Signaling Is Enhanced and TGF-beta1/SMAD2/3 Suppressed in Response to Cold Acclimation and Modulated by Thyroid Status in Hairless SHR(M). Biomedicines 2022, 10, 1707. [Google Scholar] [CrossRef]
- Decrock, E.; Hoorelbeke, D.; Ramadan, R.; Delvaeye, T.; De Bock, M.; Wang, N.; Krysko, D.V.; Baatout, S.; Bultynck, G.; Aerts, A.; et al. Calcium, oxidative stress and connexin channels, a harmonious orchestra directing the response to radiotherapy treatment? Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 1099–1120. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [Green Version]
- Arzt, M.; Drzymalski, M.A.; Ripfel, S.; Meindl, S.; Biedermann, A.; Durczok, M.; Keller, K.; Mustroph, J.; Katz, S.; Tafelmeier, M.; et al. Enhanced Cardiac CaMKII Oxidation and CaMKII-Dependent SR Ca Leak in Patients with Sleep-Disordered Breathing. Antioxidants 2022, 11, 331. [Google Scholar] [CrossRef]
- Lebek, S.; Hegner, P.; Schach, C.; Reuthner, K.; Tafelmeier, M.; Maier, L.S.; Arzt, M.; Wagner, S. A novel mouse model of obstructive sleep apnea by bulking agent-induced tongue enlargement results in left ventricular contractile dysfunction. PLoS ONE 2020, 15, e0243844. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Guan, X.; Luczak, E.D.; Lang, D.; Kutschke, W.; Gao, Z.; Yang, J.; Glynn, P.; Sossalla, S.; Swaminathan, P.D.; et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Invest. 2013, 123, 1262–1274. [Google Scholar] [CrossRef] [Green Version]
- Backs, J.; Backs, T.; Neef, S.; Kreusser, M.M.; Lehmann, L.H.; Patrick, D.M.; Grueter, C.E.; Qi, X.; Richardson, J.A.; Hill, J.A.; et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. USA 2009, 106, 2342–2347. [Google Scholar] [CrossRef]
- Mustroph, J.; Wagemann, O.; Lebek, S.; Tarnowski, D.; Ackermann, J.; Drzymalski, M.; Pabel, S.; Schmid, C.; Wagner, S.; Sossalla, S.; et al. SR Ca2+-leak and disordered excitation-contraction coupling as the basis for arrhythmogenic and negative inotropic effects of acute ethanol exposure. J. Mol. Cell. Cardiol. 2018, 116, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebek, S.; Plößl, A.; Baier, M.; Mustroph, J.; Tarnowski, D.; Lucht, C.M.; Schopka, S.; Florchinger, B.; Schmid, C.; Zausig, Y.; et al. The novel CaMKII inhibitor GS-680 reduces diastolic SR Ca leak and prevents CaMKII-dependent pro-arrhythmic activity. J. Mol. Cell. Cardiol. 2018, 118, 159–168. [Google Scholar] [CrossRef]
- Fischer, T.H.; Neef, S.; Maier, L.S. The Ca-calmodulin dependent kinase II: A promising target for future antiarrhythmic therapies? J. Mol. Cell. Cardiol. 2013, 58, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Maier, L.S.; Zhang, T.; Chen, L.; DeSantiago, J.; Brown, J.H.; Bers, D.M. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: Reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 2003, 92, 904–911. [Google Scholar] [CrossRef] [Green Version]
- Gami, A.S.; Pressman, G.; Caples, S.M.; Kanagala, R.; Gard, J.J.; Davison, D.E.; Malouf, J.F.; Ammash, N.M.; Friedman, P.A.; Somers, V.K. Association of atrial fibrillation and obstructive sleep apnea. Circulation 2004, 110, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Lebek, S.; Hegner, P.; Hultsch, R.; Rohde, J.; Rupprecht, L.; Schmid, C.; Sossalla, S.; Maier, L.S.; Arzt, M.; Wagner, S. Voltage-Gated Sodium Channel Na(V)1.8 Dysregulates Na and Ca, Leading to Arrhythmias in Patients with Sleep-Disordered Breathing. Am. J. Respir. Crit. Care Med. 2022, 206, 1428–1431. [Google Scholar] [CrossRef] [PubMed]
- Hegner, P.; Lebek, S.; Tafelmeier, M.; Camboni, D.; Schopka, S.; Schmid, C.; Maier, L.S.; Arzt, M.; Wagner, S. Sleep-disordered breathing is independently associated with reduced atrial connexin 43 expression. Heart Rhythm 2021, 18, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Neef, S.; Dybkova, N.; Sossalla, S.; Ort, K.R.; Fluschnik, N.; Neumann, K.; Seipelt, R.; Schondube, F.A.; Hasenfuss, G.; Maier, L.S. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 2010, 106, 1134–1144. [Google Scholar] [CrossRef] [Green Version]
- Voigt, N.; Li, N.; Wang, Q.; Wang, W.; Trafford, A.W.; Abu-Taha, I.; Sun, Q.; Wieland, T.; Ravens, U.; Nattel, S.; et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012, 125, 2059–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassal, D.; Gratz, D.; Hund, T.J. Challenges and Opportunities for Therapeutic Targeting of Calmodulin Kinase II in Heart. Front Pharmacol. 2020, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Rossi, V.A.; Stradling, J.R.; Kohler, M. Effects of obstructive sleep apnoea on heart rhythm. Eur. Respir. J. 2013, 41, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Ruff, H.M.; Weber, S.L.; Bellmann, S.; Sowa, T.; Schulte, T.; Anderson, M.E.; Grandi, E.; Bers, D.M.; Backs, J.; et al. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 2011, 108, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dybkova, N.; Wagner, S.; Backs, J.; Hund, T.J.; Mohler, P.J.; Sowa, T.; Nikolaev, V.O.; Maier, L.S. Tubulin polymerization disrupts cardiac beta-adrenergic regulation of late INa. Cardiovasc. Res. 2014, 103, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linz, D.; Schotten, U.; Neuberger, H.R.; Bohm, M.; Wirth, K. Negative tracheal pressure during obstructive respiratory events promotes atrial fibrillation by vagal activation. Heart Rhythm 2011, 8, 1436–1443. [Google Scholar] [CrossRef]
- Toischer, K.; Rokita, A.G.; Unsold, B.; Zhu, W.; Kararigas, G.; Sossalla, S.; Reuter, S.P.; Becker, A.; Teucher, N.; Seidler, T.; et al. Differential cardiac remodeling in preload versus afterload. Circulation 2010, 122, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Costanzo, M.R.; Khayat, R.; Ponikowski, P.; Augostini, R.; Stellbrink, C.; Mianulli, M.; Abraham, W.T. Mechanisms and clinical consequences of untreated central sleep apnea in heart failure. J. Am. Coll. Cardiol. 2015, 65, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Cowie, M.R.; Linz, D.; Redline, S.; Somers, V.K.; Simonds, A.K. Sleep Disordered Breathing and Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 608–624. [Google Scholar] [CrossRef]
- Lavie, L.; Lavie, P. Molecular mechanisms of cardiovascular disease in OSAHS: The oxidative stress link. Eur. Respir. J. 2009, 33, 1467–1484. [Google Scholar] [CrossRef]
- Corbi, G.; Conti, V.; Russomanno, G.; Longobardi, G.; Furgi, G.; Filippelli, A.; Ferrara, N. Adrenergic signaling and oxidative stress: A role for sirtuins? Front Physiol. 2013, 4, 324. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zhang, T.; Ginsburg, K.S.; Mishra, S.; Brown, J.H.; Bers, D.M. CaMKIIdeltaC slows [Ca]i decline in cardiac myocytes by promoting Ca sparks. Biophys. J. 2012, 102, 2461–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegner, P.; Drzymalski, M.; Biedermann, A.; Memmel, B.; Durczok, M.; Wester, M.; Floerchinger, B.; Provaznik, Z.; Schmid, C.; Zausig, Y.; et al. SAR296968, a Novel Selective Na+/Ca2+ Exchanger Inhibitor, Improves Ca2+ Handling and Contractile Function in Human Atrial Cardiomyocytes. Biomedicines 2022, 10, 1932. [Google Scholar] [CrossRef]
- Traaen, G.M.; Aakeroy, L.; Hunt, T.E.; Overland, B.; Bendz, C.; Sande, L.O.; Aakhus, S.; Fagerland, M.W.; Steinshamn, S.; Anfinsen, O.G.; et al. Effect of Continuous Positive Airway Pressure on Arrhythmia in Atrial Fibrillation and Sleep Apnea: A Randomized Controlled Trial. Am. J. Respir. Crit. Care Med. 2021, 204, 573–582. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, R.D.; Antic, N.A.; Heeley, E.; Luo, Y.; Ou, Q.; Zhang, X.; Mediano, O.; Chen, R.; Drager, L.F.; Liu, Z.; et al. CPAP for Prevention of Cardiovascular Events in Obstructive Sleep Apnea. N. Engl. J. Med. 2016, 375, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Arzt, M.; Young, T.; Finn, L.; Skatrud, J.B.; Ryan, C.M.; Newton, G.E.; Mak, S.; Parker, J.D.; Floras, J.S.; Bradley, T.D. Sleepiness and sleep in patients with both systolic heart failure and obstructive sleep apnea. Arch. Intern. Med. 2006, 166, 1716–1722. [Google Scholar] [CrossRef] [Green Version]
- Cowie, M.R.; Woehrle, H.; Wegscheider, K.; Angermann, C.; d’Ortho, M.P.; Erdmann, E.; Levy, P.; Simonds, A.K.; Somers, V.K.; Zannad, F.; et al. Adaptive Servo-Ventilation for Central Sleep Apnea in Systolic Heart Failure. N. Engl. J. Med. 2015, 373, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- De Greef, Y.; Stroker, E.; Schwagten, B.; Kupics, K.; De Cocker, J.; Chierchia, G.B.; de Asmundis, C.; Stockman, D.; Buysschaert, I. Complications of pulmonary vein isolation in atrial fibrillation: Predictors and comparison between four different ablation techniques: Results from the MIddelheim PVI-registry. Europace 2018, 20, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomstrom-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Neef, S.; Mann, C.; Zwenger, A.; Dybkova, N.; Maier, L.S. Reduction of SR Ca2+ leak and arrhythmogenic cellular correlates by SMP-114, a novel CaMKII inhibitor with oral bioavailability. Basic Res. Cardiol. 2017, 112, 45. [Google Scholar] [CrossRef] [PubMed]
- Neef, S.; Steffens, A.; Pellicena, P.; Mustroph, J.; Lebek, S.; Ort, K.R.; Schulman, H.; Maier, L.S. Improvement of cardiomyocyte function by a novel pyrimidine-based CaMKII-inhibitor. J. Mol. Cell. Cardiol. 2018, 115, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellicena, P.; Schulman, H. CaMKII inhibitors: From research tools to therapeutic agents. Front Pharmacol. 2014, 5, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauverger, P.; Ozoux, M.L.; Begis, G.; Glenat, V.; Briand, V.; Philippo, M.C.; Daveu, C.; Tavares, G.; Roy, S.; Corbier, A.; et al. Reversion of cardiac dysfunction by a novel orally available calcium/calmodulin-dependent protein kinase II inhibitor, RA306, in a genetic model of dilated cardiomyopathy. Cardiovasc. Res. 2020, 116, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Mustroph, J.; Drzymalski, M.; Baier, M.; Pabel, S.; Biedermann, A.; Memmel, B.; Durczok, M.; Neef, S.; Sag, C.M.; Floerchinger, B.; et al. The oral Ca/calmodulin-dependent kinase II inhibitor RA608 improves contractile function and prevents arrhythmias in heart failure. ESC Heart Fail. 2020, 7, 2871–2883. [Google Scholar] [CrossRef]
- Hegner, P.; Lebek, S.; Maier, L.S.; Arzt, M.; Wagner, S. The Effect of Gender and Sex Hormones on Cardiovascular Disease, Heart Failure, Diabetes, and Atrial Fibrillation in Sleep Apnea. Front Physiol. 2021, 12, 741896. [Google Scholar] [CrossRef]
- Lebek, S.; Chemello, F.; Caravia, X.M.; Tan, W.; Li, H.; Chen, K.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Ablation of CaMKIIdelta oxidation by CRISPR-Cas9 base editing as a therapy for cardiac disease. Science 2023, 379, 179–185. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Mesarwi, O.A.; Sharma, E.V.; Jun, J.C.; Polotsky, V.Y. Metabolic dysfunction in obstructive sleep apnea: A critical examination of underlying mechanisms. Sleep Biol. Rhythm. 2015, 13, 2–17. [Google Scholar] [CrossRef] [Green Version]
- Levy, P.; Bonsignore, M.R.; Eckel, J. Sleep, sleep-disordered breathing and metabolic consequences. Eur. Respir. J. 2009, 34, 243–260. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hegner, P.; Lebek, S.; Schaner, B.; Ofner, F.; Gugg, M.; Maier, L.S.; Arzt, M.; Wagner, S. CaMKII-Dependent Contractile Dysfunction and Pro-Arrhythmic Activity in a Mouse Model of Obstructive Sleep Apnea. Antioxidants 2023, 12, 315. https://doi.org/10.3390/antiox12020315
Hegner P, Lebek S, Schaner B, Ofner F, Gugg M, Maier LS, Arzt M, Wagner S. CaMKII-Dependent Contractile Dysfunction and Pro-Arrhythmic Activity in a Mouse Model of Obstructive Sleep Apnea. Antioxidants. 2023; 12(2):315. https://doi.org/10.3390/antiox12020315
Chicago/Turabian StyleHegner, Philipp, Simon Lebek, Benedikt Schaner, Florian Ofner, Mathias Gugg, Lars Siegfried Maier, Michael Arzt, and Stefan Wagner. 2023. "CaMKII-Dependent Contractile Dysfunction and Pro-Arrhythmic Activity in a Mouse Model of Obstructive Sleep Apnea" Antioxidants 12, no. 2: 315. https://doi.org/10.3390/antiox12020315
APA StyleHegner, P., Lebek, S., Schaner, B., Ofner, F., Gugg, M., Maier, L. S., Arzt, M., & Wagner, S. (2023). CaMKII-Dependent Contractile Dysfunction and Pro-Arrhythmic Activity in a Mouse Model of Obstructive Sleep Apnea. Antioxidants, 12(2), 315. https://doi.org/10.3390/antiox12020315