


NLRX1 Inhibits LPS-Induced Microglial Death via Inducing p62-Dependent HO-1 Expression, Inhibiting MLKL and Activating PARP-1

 , ,
, , 
Abstract
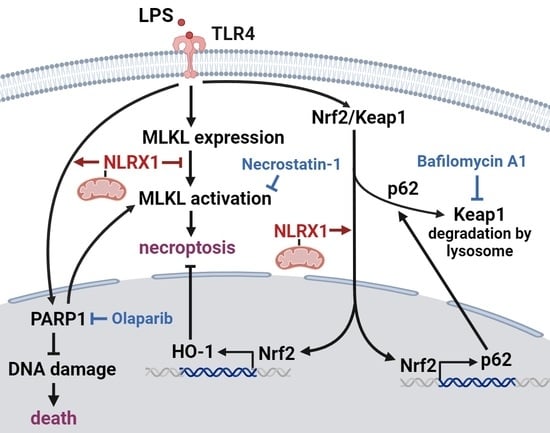
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Generation of shRNA and siRNA Knockdown Cell Lines
2.4. Annexin V/PI Staining
2.5. Measurement of Intracellular ROS Production
2.6. Immunoblotting
2.7. Reverse-Transcription and Real-Time Polymerase Chain Reaction (RT-PCR)
2.8. Confocal Microscopy
2.9. Quantification and Statistical Analysis
3. Results
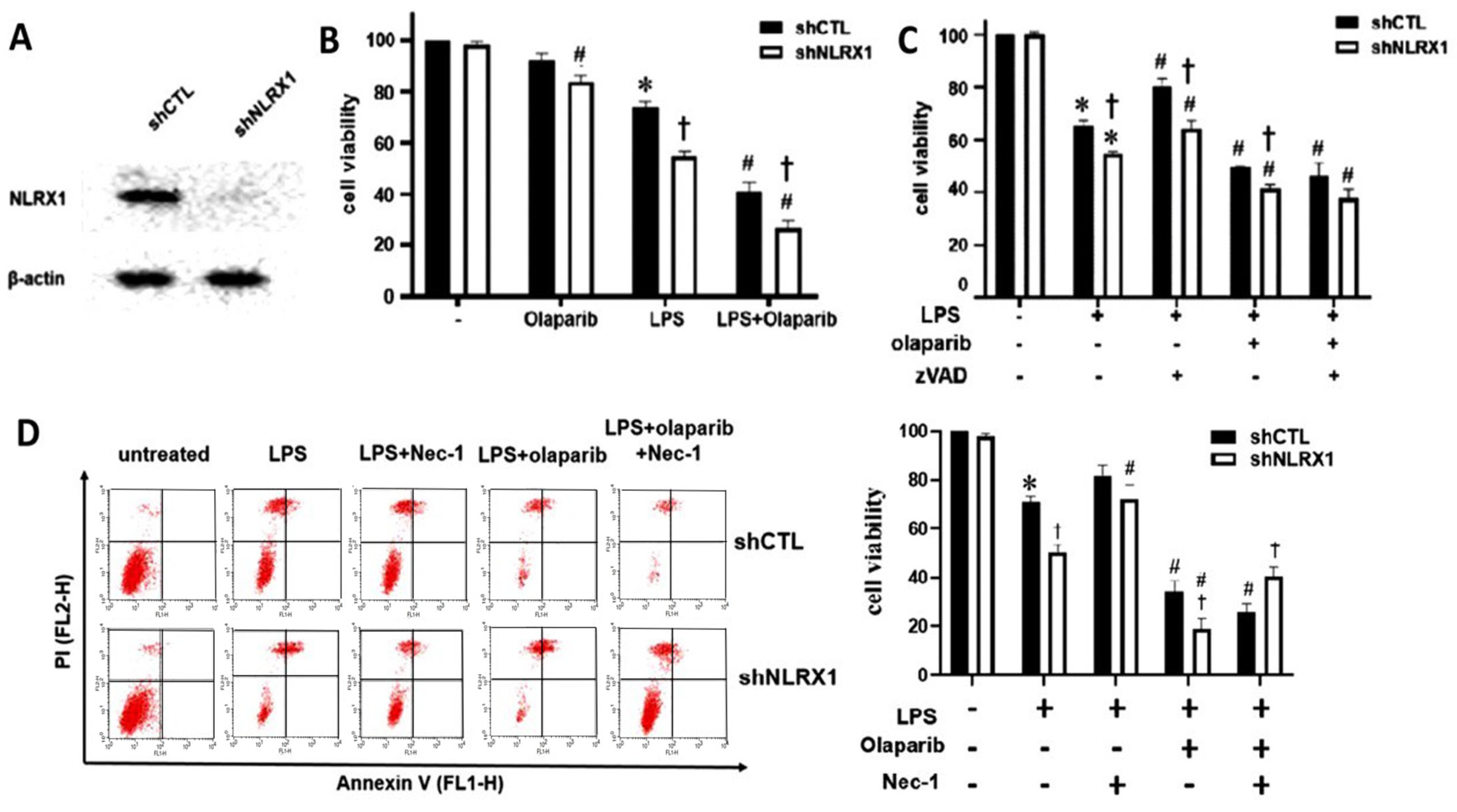
3.1. NLRX1 Silencing Enhances LPS-Induced Mixed-Type Cell Death in SM826 Cells, Which Is Additive to the PARP-1 Inhibitor
3.2. NLRX1 Silencing Inhibits LPS-Induced PARP-1 Activation and DNA Repair
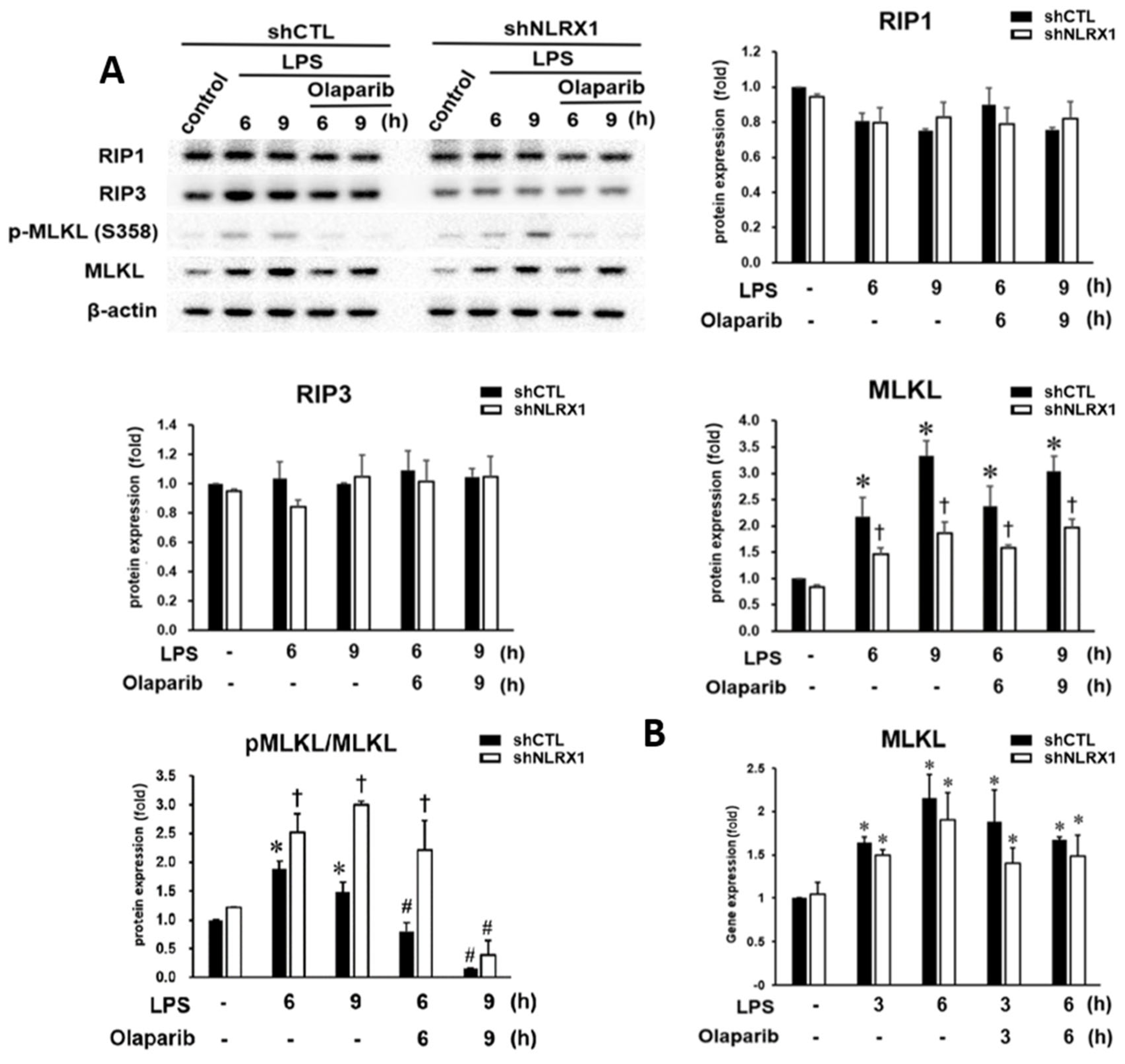
3.3. NLRX1 Silencing Enhances MLKL Activation
3.4. NLRX1 Silencing Reduces LPS-Induced HO-1 Expression
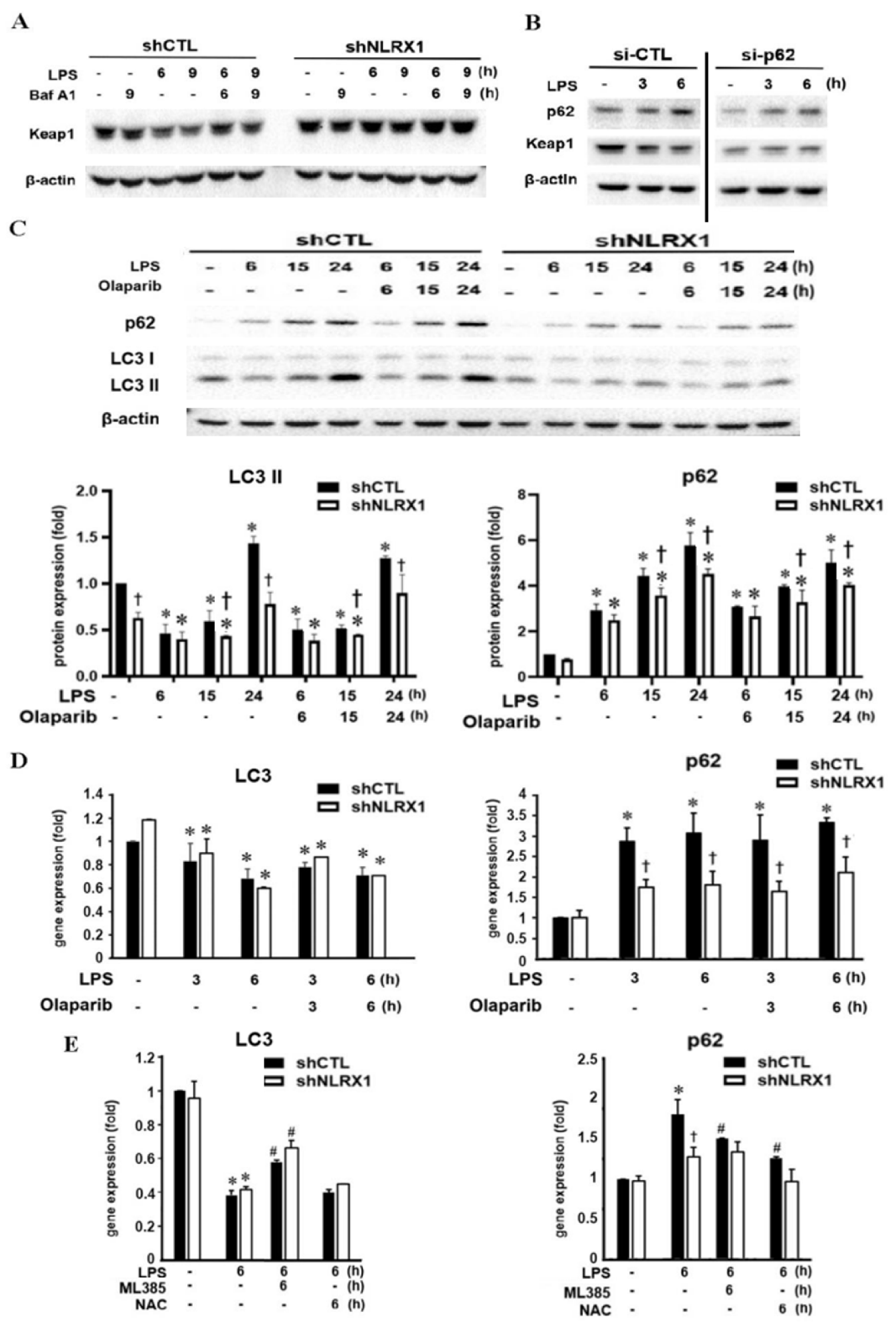
3.5. NLRX1 Silencing Decreases the LPS-Induced Nrf2/HO-1 Pathway via Inhibiting Keap1 Downregulation
3.6. NLRX1 Silencing Suppresses LPS-Induced Autophagy and p62 Upregulation
3.7. NLRX1 Silencing Inhibits PARP-1 Activation Independently of Autophagy Suppression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DAPI | 4′,6-diamidino-2-phenylindole |
| DCFDA | Dichlorodihydrofluorescein diacetate |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | Fetal bovine serum |
| H2O2 | Hydrogen peroxide |
| HO-1 | Heme oxygenase-1 |
| IFN | Interferon |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LPS | Lipopolysaccharide |
| MAVS | Mitochondrial antiviral-signaling protein |
| MLKL | Mixed lineage kinase domain-like pseudokinase |
| NAC | N-acetyl-L-cysteine |
| NLR | NOD-like receptor |
| NLRX1 | Nucleotide-binding oligomerization domain, leucine-rich repeat-containing X1 |
| PAR | Poly (ADP-ribose) |
| PARP-1 | Poly (ADP-ribose) polymerase-1 |
| PBS | Phosphate buffered saline |
| PI | Propidium iodide |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase 1 |
| ROS | Reactive oxygen species |
| RT-PCR | Reverse-transcription and real-time polymerase chain reaction |
| SDS | Sodium dodecyl sulfate |
| TBST | Tris-buffer saline with Tween 20 |
| ZnPP | Zinc protoporphyrin |
References
- Sekar, P.; Huang, D.Y.; Hsieh, S.L.; Chang, S.F.; Lin, W.W. AMPK-dependent and independent actions of P2X7 in regulation of mitochondrial and lysosomal functions in microglia. Cell Commun. Signal 2018, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Hagan, N.; Kane, J.L.; Grover, D.; Woodworth, L.; Madore, C.; Saleh, J.; Sancho, J.; Liu, J.; Li, Y.; Proto, J.; et al. CSF1R signaling is a regulator of pathogenesis in progressive MS. Cell Death Dis. 2020, 11, 904. [Google Scholar] [CrossRef] [PubMed]
- Sekar, P.; Hsiao, G.; Chen, Y.S.; Lin, W.W.; Chan, C.M. P2X7 is involved in the mouse retinal degeneration via the coordinated actions in different retinal cell types. Antioxidants 2023, 12, 141. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Son, S.M.; Mook-Jung, I. Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia 2016, 64, 2274–2290. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.K.; Zelic, M.; Han, Y.; Teeple, E.; Chen, L.; Sadeghi, M.; Shankara, S.; Guo, L.; Li, C.; Pontarelli, F.; et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat. Neurosci. 2021, 26, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Wang, J.; Kawanokuchi, J.; Mitsuma, N.; Mizuno, T.; Suzumura, A. Interferon-γ induces microglial-activation-induced cell death: A hypothetical mechanism of relapse and remission in multiple sclerosis. Neurobiol. Dis. 2006, 22, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Li, J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013, 4, e716. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yang, M.S.; Jiang, J.; Ganesh, T.; Joe, E.; Dingledine, R. EP2 receptor signaling regulates microglia death. Mol. Pharmacol. 2015, 88, 161–170. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, T.; Sun, X.; Zheng, Y.; Cheng, B.; Li, M.; Liu, X.; He, C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2017, 25, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.G.; Mohney, R.P.; Wilson, B.; Jeohn, G.H.; Liu, B.; Hong, J.S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, K.; Gao, H.M.; Mandavilli, B.; Wang, J.Y.; Hong, J.S. Molecular consequences of activated microglia in the brain: Overactivation induces apoptosis. J. Neurochem. 2001, 77, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, P.; Shrivastava, P.; Singh, S.M.; Sodhi, A.; Heintz, N.H. Baculovirus p35 inhibits NO-induced apoptosis in activated macrophages by inhibiting cytochrome c release. J. Cell Sci. 2004, 117, 3031–3039. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.Y.; Lee, H.; Jung, B.Y.; Ock, J.; Lee, M.S.; Lee, W.H.; Suk, K. TLR4, but not TLR2, signals autoregulatory apoptosis of cultured microglia: A critical role of IFN-β as a decision maker. J. Immunol. 2005, 174, 6467–6476. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.S.; Chien, C.C.; Cheng, K.T.; Subbaraju, G.V.; Chen, Y.C. Hispolon suppresses LPS- or LTA-induced iNOS/NO production and apoptosis in BV-2 microglial cells. Am. J. Chin. Med. 2017, 45, 1649–1666. [Google Scholar] [CrossRef] [PubMed]
- Pislar, A.; Nedeljkovic, B.B.; Peric, M.; Jakos, T.; Zidar, N.; Kos, J. Cysteine peptidase cathepsin X as a therapeutic target for simultaneous TLR3/4-mediated microglia activation. Mol. Neurobiol. 2022, 59, 2258–2276. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.R.; Dionísio, P.A.; Brito, H.; Franco, L.; Rodrigues, C.A.; Guedes, R.C.; Afonso, C.A.; Amaral, J.D.; Rodrigues, C.M. Phenotypic screening identifies a new oxazolone inhibitor of necroptosis and neuroinflammation. Cell Death Discov. 2018, 4, 65. [Google Scholar] [CrossRef]
- Dionísio, P.E.A.; Oliveira, S.R.; Amaral, J.S.J.D.; Rodrigues, C.M.P. Loss of microglial Parkin inhibits necroptosis and contributes to neuroinflammation. Mol. Neurobiol. 2019, 56, 2990–3004. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Liu, X.; Chen, C.; Zhang, X.; Xie, P.; Liu, Y.; Zhou, S.; Tang, J. Erlotinib protests against LPS-induced parthanatos through inhibiting macrophage surface TLR4 expression. Cell Death Discov. 2021, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Snaka, T.; Fasel, N. Behind the scenes: Nod-like receptor X1 controls inflammation and metabolism. Front. Cell. Infect. Microbiol. 2020, 10, 609812. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Soares, F.; Tattoli, I.; Castanier, C.; Philpott, D.J.; Stephen, E.; Girardin, S.E. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J. Cell Sci. 2009, 122, 3161–3168. [Google Scholar] [CrossRef] [PubMed]
- Fekete, T.; Bencze, D.; Bíró, E.; Benkő, S.; Pázmándi, K. Focusing on the cell type specific regulatory actions of NLRX1. Int. J. Mol. Sci. 2021, 22, 1316. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Moore, C.B.; Schneider, M.; Lei, Y.; Davis, B.K.; Scull, M.A.; Gris, D.; Roney, K.E.; Zimmermann, A.G.; Bowzard, J.B.; et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity 2011, 34, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Imbeault, E.; Mahvelati, T.M.; Braun, R.; Gris, P.; Gris, D. Nlrx1 regulates neuronal cell death. Mol. Brain 2014, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Poteryakhina, A.; Zheltukhin, A.; Bhatelia, K.; Prajapati, P.; Sripada, L.; Tomar, D.; Singh, R.; Singh, A.K.; Chumakov, P.M.; et al. NLRX1 acts as tumor suppressor by regulating TNF-α induced apoptosis and metabolism in cancer cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Zhao, Y.; She, J.; Zhu, Y.; Zhao, Y.; Liu, L.; Zhang, Y. NLRX1 alleviates lipopolysaccharide-induced apoptosis and inflammation in chondrocytes by suppressing the activation of NF-κB signaling. Int. Immunopharmacol. 2019, 71, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Theus, M.H.; Brickler, T.; Meza, A.L.; Coutermarsh-Ott, S.; Hazy, A.; Gris, D.; Allen, I. Loss of NLRX1 exacerbates neural tissue damage and NF-κB signaling following brain injury. J. Immunol. 2017, 199, 3547–3558. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Amrani, A.; Gris, D. NLRX1 enhances glutamate uptake and inhibits glutamate release by astrocytes. Cells 2019, 8, 400. [Google Scholar] [CrossRef]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, 6414. [Google Scholar] [CrossRef] [PubMed]
- Stoica, B.A.; Loane, D.J.; Zhao, Z.; Kabadi, S.V.; Hanscom, M.; Byrnes, K.R.; Faden, A.I. PARP-1 inhibition attenuates neuronal loss, microglia activation and neurological deficits after traumatic brain injury. J. Neurotrauma 2014, 31, 758–772. [Google Scholar] [CrossRef]
- Park, H.; Kam, T.I.; Dawson, T.M.; Dawson, V.L. Poly (ADP-ribose) (PAR)-dependent cell death in neurodegenerative diseases. Int. Rev. Cell Mol. Biol. 2020, 353, 1–29. [Google Scholar] [PubMed]
- Wang, Y.; Pleasure, D.; Deng, W.; Guo, F. Therapeutic potentials of poly (ADP-Ribose) polymerase 1 (PARP1) inhibition in multiple sclerosis and animal models: Concept revisiting. Adv. Sci. 2022, 9, 2102853. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Yang, C.N.; Pan, H.C.; Sung, Y.J.; Liao, K.K.; Chen, W.B.; Lin, W.Z.; Sheu, M.L. IL-13 downregulates PPAR-γ/heme oxygenase-1 via ER stress-stimulated calpain activation: Aggravation of activated microglia death. Cell. Mol. Life Sci. 2010, 67, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Lin, C.H.; Hsu, P.C.; Sun, Y.Y.; Huang, Y.J.; Zhuo, J.H.; Wang, C.Y.; Gan, Y.L.; Hung, C.C.; Kuan, C.Y.; et al. Aryl hydrocarbon receptor mediates both proinflammatory and anti-inflammatory effects in lipopolysaccharide-activated microglia. Glia 2015, 63, 1138–1154. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Jiang, N.; Su, W.; Zhuo, Y. Necroptosis: A novel pathway in neuroinflammation. Front. Pharmacol. 2021, 12, 701564. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Chuang, W.C.; Kuang, H.N.; Cheng, C.Y.; Huang, D.Y.; Lin, W.W. Pan-caspase inhibitor zVAD induces necroptosis and autophagic cell death in TLR3/4-stimulated macrophages. Mol. Cells 2022, 45, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.Y.; Huang, D.Y.; Lin, W.W. PARP-1 regulates inflammasome activity by poly-ADP-ribosylation of NLRP3 and interaction with TXNIP in primary macrophages. Cell. Mol. Life Sci. 2022, 79, 108. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.J.; Fischman, D.A.; Hammerling, U. Vitamin A depletion causes oxidative stress, mitochondrial dysfunction, and PARP-1-dependent energy deprivation. FASEB J. 2008, 22, 3878–3887. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.Y.; Wu, N.L.; Hung, C.F.; Bai, P.; Dai, Y.S.; Lin, W.W. PARP-1 involves in UVB-induced inflammatory response in keratinocytes and skin injury via regulation of ROS-dependent EGFR transactivation and p38 signaling. FASEB J. 2021, 35, 21393. [Google Scholar] [CrossRef]
- Diaz-Hernandez, J.I.; Moncada, S.; Bolaños, J.P.; Almeida, A. Poly (ADP-ribose) polymerase-1 protects neurons against apoptosis induced by oxidative stress. Cell Death Differ. 2007, 14, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Cheong, K.J.H.; Huang, D.Y.; Sekar, P.; Chen, R.J.; Cheng, I.H.J.; Chan, C.M.; Chen, Y.S.; Lin, W.W. CASK mediates oxidative stress-induced microglial apoptosis-inducing factor-independent parthanatos cell death via promoting PARP-1 hyperactivation and mitochondrial dysfunction. Antioxidants 2024, 13, 343. [Google Scholar] [CrossRef] [PubMed]
- Schallner, N.; Pandit, R.; LeBlanc, R.; Thomas, A.J.; Ogilvy, C.S.; Zuckerbraun, B.S.; Gallo, D.; Otterbein, L.E.; Hanafy, K.A. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Investig. 2015, 125, 2609–2625. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.Y.; Chen, Z.; Jasmer, K.J.; Chuang, D.Y.; Gu, Z.; Hannink, M.; Simonui, A. Quercetin attenuates inflammatory responses in BV-2 microglial cells: Role of MAPKs on the Nrf2 pathway and induction of heme oxygenase-1. PLoS ONE 2015, 10, e0141509. [Google Scholar] [CrossRef] [PubMed]
- Prawan, A.; Kundu, J.K.; Surh, Y.J. Molecular basis of heme oxygenase-1 induction: Implications for chemoprevention and chemoprotection. Antioxid. Redox Signal. 2009, 7, 1688–1703. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Zandieh, M.A.; Ziaolhagh, S.; Mojtabavi, S.; Sadi, F.H.; Koohpar, Z.K.; Ghanbarirad, M.; Haghighatfard, A.; Behroozaghdam, M.; Khorrami, R.; et al. Nrf2 signaling in diabetic nephropathy, cardiomyopathy and neuropathy: Therapeutic targeting, challenges and future prospective. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2023, 1869, 166714. [Google Scholar] [CrossRef] [PubMed]
- Sani, G.; Margoni, S.; Brugnami, A.; Ferrara, O.M.; Bernardi, E.; Simonetti, A.; Monti, L.; Mazza, M.; Janiri, D.; Moccia, L.; et al. The Nrf2 pathway in depressive disorders: A systematic review of animal and human studies. Antioxidants 2023, 12, 817. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Wang, Z.; Fu, Z.; Ma, H.; Jiang, M.; Xu, A.; Zhang, W. p62/SQSTM1 protects against cisplatin-induced oxidative stress in kidneys by mediating the cross talk between autophagy and the Keap1-Nrf2 signaling pathway. Free Radic. Res. 2019, 53, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Essandoh, K.; Wang, X.; Li, Y.; Huang, W.; Chen, J.; Peng, J.; Jiang, D.S.; Mu, X.; Wang, C.; et al. Tsg101 positively regulates p62-Keap1-Nrf2 pathway to protect hearts against oxidative damage. Redox Biol. 2020, 32, 101453. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Park, J.S.; Lee, Y.S.; Han, J.; Lee, D.K.; Kwon, S.W.; Han, D.H.; Lee, Y.H.; Bae, S.H. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy 2020, 16, 1949–1973. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Harder, B.; Rojo De La Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88 Pt B, 199–204. [Google Scholar] [CrossRef]
- Mao, K.; Chen, J.; Yu, H.; Li, H.; Ren, Y.; Wu, X.; Wen, Y.; Zou, F.; Li, W. Poly (ADP-ribose) polymerase 1 inhibition prevents neurodegeneration and promotes α-synuclein degradation via transcription factor EB-dependent autophagy in mutant α-synucleinA53T model of Parkinson’s disease. Aging Cell 2020, 19, e13163. [Google Scholar] [CrossRef]
- Rodríguez-Vargas, J.M.; Oliver-Pozo, F.J.; Dantzer, F. PARP1 and poly(ADP-ribosyl)ation signaling during autophagy in response to nutrient deprivation. Oxidative Med. Cell Longev. 2019, 2019, 2641712. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.Y.T.; Sekar, P.; Huang, D.Y.; Hsu, S.H.; Chan, C.M.; Lin, W.W. Spatiotemporal roles of AMPK in PARP-1- and autophagy-dependent retinal pigment epithelial cell death caused by UVA. J. Biomed. Sci. 2023, 30, 91. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.I.; Maeda, D.; Xiao, Q.; Srinivasula, S.M. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Liu, L.; Ren, F.; Gao, Q.; Xu, S.; Hou, B.; Wang, Y.; Jiang, X.; Che, Y. Sunitinib induces genomic instability of renal carcinoma cells through affecting the interaction of LC3-II and PARP-1. Cell Death Dis. 2017, 8, e2988. [Google Scholar] [CrossRef] [PubMed]
- Gharagozloo, M.; Gris, K.V.; Mahvelati, T.; Amrani, A.; Lukens, J.R.; Gris, D. NLR-dependent regulation of inflammation in multiple sclerosis. Front. Immunol. 2018, 8, 2012. [Google Scholar] [PubMed]
- Liu, M.; Liu, K.; Cheng, D.; Zheng, B.; Li, S.; Mo, Z. The regulatory role of NLRX1 in innate immunity and human disease. Cytokine 2022, 160, 156055. [Google Scholar] [CrossRef] [PubMed]
- Pickering, R.J.; Booty, L.M. NLR in eXile: Emerging roles of NLRX1 in immunity and human disease. Immunology 2021, 162, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhou, Z.M.; Zhu, L.; Chen, L.; Jiang, Z.L.; Wu, X.T. Underlying mechanisms and related diseases behind the complex regulatory role of NOD-like receptor X1. DNA Cell Biol. 2022, 41, 469–478. [Google Scholar] [CrossRef]
- Li, S.; Zhou, Y.; Gu, X.; Zhang, X.; Zhongzhi, J. NLRX1/FUNDC1/NIPSNAP1-2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 2021, 54, e12986. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, S.; Li, F.; Qin, L. NLRX1 attenuates apoptosis and inflammatory responses in myocardial ischemia by inhibiting MAVS-dependent NLRP3 inflammasome activation. Mol. Immunol. 2016, 76, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Philipson, C.W.; Bassaganya-Riera, J.; Viladomiu, M.; Kronsteiner, B.; Abedi, V.; Hoops, S.; Michalak, P.; Kang, L.; Girardin, S.E.; Hontecillas, R. Modeling the regulatory mechanisms by which NLRX1 modulates innate immune responses to Helicobacter pylori infection. PLoS ONE 2015, 10, e0137839. [Google Scholar] [CrossRef]
- Stokman, G.; Kors, L.; Bakker, P.J.; Rampanelli, E.; Claessen, N.; Teske, G.J.; Butter, L.; van Andel, H.; van den Bergh Weerman, M.A.; Larsen, P.W.; et al. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J. Exp. Med. 2017, 214, 2405–2420. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Yoon, C.M.; Kim, B.H.; Lee, C.M.; Zhou, Y.; Sauler, M.; Homer, R.; Dhamija, A.; Boffa, D.; West, A.P.; et al. Suppression of NLRX1 in chronic obstructive pulmonary disease. J. Clin. Investig. 2015, 125, 2458–2462. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Nam, H.; Kim, L.E.; Jeon, Y.; Min, H.; Ha, S.; Lee, Y.; Kim, S.Y.; Lee, S.J.; Kim, E.K.; et al. TLR4 (toll-like receptor 4) activation suppresses autophagy through inhibition of FOXO3 and impairs phagocytic capacity of microglia. Autophagy 2019, 15, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhu, M.; Che, X.; Wang, H.; Liang, X.J.; Wu, C.; Xue, X.; Yang, J. Lipopolysaccharide induces neuroinflammation in microglia by activating the MTOR pathway and downregulating Vps34 to inhibit autophagosome formation. J. Neuroinflammation 2020, 17, 18. [Google Scholar] [CrossRef]
- Saito, Y.; Yako, T.; Otsu, W.; Nakamura, S.; Inoue, Y.; Muramatsu, A.; Nakagami, Y.; Shimazawa, M.; Hara, H. A triterpenoid Nrf2 activator, RS9, promotes LC3-associated phagocytosis of photoreceptor outer segments in a p62-independent manner. Free Radic. Biol. Med. 2020, 152, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Hang, S.; Zhang, W.; Mao, C.; Li, D. An update on the bridging factors connecting autophagy and Nrf2 antioxidant pathway. Front. Cell Dev. Biol. 2023, 11, 1232241. [Google Scholar] [CrossRef]
- Liu, L.; Li, J.; Ke, Y.; Zeng, X.; Gao, J.; Ba, X.; Wang, R. The key players of parthanatos: Opportunities for targeting multiple levels in the therapy of parthanatos-based pathogenesis. Cell. Mol. Life Sci. 2022, 79, 60. [Google Scholar] [CrossRef]
- Park, J.M.; Tougeron, D.; Huang, S.; Okamoto, K.; Sinicrope, F.A. Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PLoS ONE 2014, 9, 100819. [Google Scholar]
- Zhao, Z.; Oh, S.; Li, D.; Ni, D.; Pirooz, S.D.; Lee, J.H.; Yang, S.; Lee, J.Y.; Ghozalli, I.; Costanzo, V.; et al. A dual role for UVRAG in maintaining chromosomal stability independent of autophagy. Dev. Cell 2012, 22, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Weill, S.I. Heme oxgenase-1, a cardinal modulator of regulated cell death and inflammation. Cells 2021, 10, 515. [Google Scholar] [CrossRef] [PubMed]
- Kopacz, A.; Kloska, D.; Forman, H.J.; Jozkowicz, A.; Grochot-Przeczek, A. Beyond repression of Nrf2: An update on Keap1. Free Radic. Biol. Med. 2020, 157, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Li, Q.; Shen, X.; Yao, Y.; Li, L.; Ma, H. Dehydroepiandrosterone attenuates LPS-induced inflammatory responses via activation of Nrf2 in RAW264.7 macrophages. Mol. Immunol. 2021, 131, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Bi, P.Y.; Killackey, S.A.; Schweizer, L.; Girardin, S.E. NLRX1: Versatile functions of a mitochondrial NLR protein that controls mitophagy. Biomed. J. 2023, 47, 100635. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Wen, H.; Yu, Y.; Taxman, D.J.; Zhang, L.; Widman, D.G.; Swanson, K.V.; Wen, K.W.; Damania, B.; Moore, C.B.; et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 2012, 36, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Gillette, D.D.; Li, X.; Zhang, Z.; Wen, H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J. Biol. Chem. 2014, 289, 17020–17029. [Google Scholar] [CrossRef] [PubMed]
- Knuth, A.K.; Rosler, S.; Schenk, B.; Kowald, L.; Van Wijk, S.J.L.; Fulda, S. Interferons transcriptionally up-regulate MLKL expression in cancer cells. Neoplasia 2019, 21, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Tak, U.; Sun, J.; Orihuela, C.J.; Niederweis, M. NAD+ depletion triggers macrophage necroptosis, a cell death pathway exploited by Mycobacterium tuberculosis. Cell Rep. 2018, 24, 429–440. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-L.; Huang, D.-Y.; Klochkov, V.; Chan, C.-M.; Chen, Y.-S.; Lin, W.-W. NLRX1 Inhibits LPS-Induced Microglial Death via Inducing p62-Dependent HO-1 Expression, Inhibiting MLKL and Activating PARP-1. Antioxidants 2024, 13, 481. https://doi.org/10.3390/antiox13040481
Huang Y-L, Huang D-Y, Klochkov V, Chan C-M, Chen Y-S, Lin W-W. NLRX1 Inhibits LPS-Induced Microglial Death via Inducing p62-Dependent HO-1 Expression, Inhibiting MLKL and Activating PARP-1. Antioxidants. 2024; 13(4):481. https://doi.org/10.3390/antiox13040481
Chicago/Turabian StyleHuang, Yu-Ling, Duen-Yi Huang, Vladlen Klochkov, Chi-Ming Chan, Yuan-Shen Chen, and Wan-Wan Lin. 2024. "NLRX1 Inhibits LPS-Induced Microglial Death via Inducing p62-Dependent HO-1 Expression, Inhibiting MLKL and Activating PARP-1" Antioxidants 13, no. 4: 481. https://doi.org/10.3390/antiox13040481
APA StyleHuang, Y. -L., Huang, D. -Y., Klochkov, V., Chan, C. -M., Chen, Y. -S., & Lin, W. -W. (2024). NLRX1 Inhibits LPS-Induced Microglial Death via Inducing p62-Dependent HO-1 Expression, Inhibiting MLKL and Activating PARP-1. Antioxidants, 13(4), 481. https://doi.org/10.3390/antiox13040481