Differential Effects of Superoxide Dismutase Mimetics after Mechanical Overload of Articular Cartilage

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Harvest and Culture
2.2. SOD Mimetic Treatments
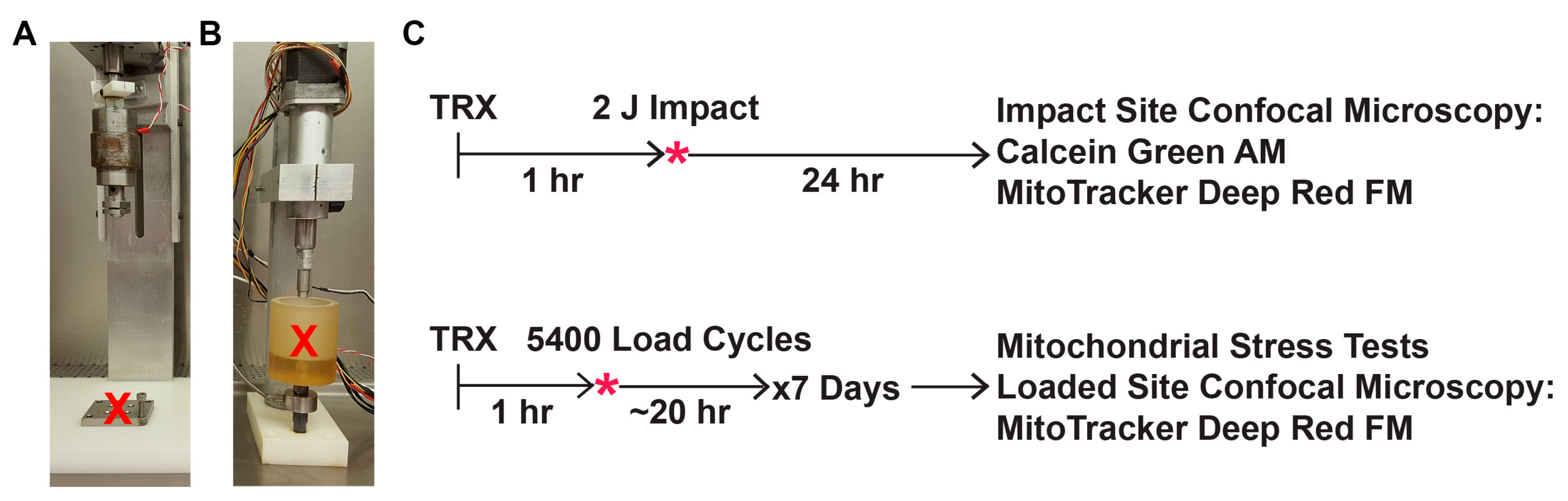
2.3. Mechanical Impact Delivered via Drop Tower
2.4. Mechanical Overload Delivered via Repeated Axial Compression
2.5. Post-Impact Oxidant Production, Cell Viability, and Mitochondrial Content
2.6. Mitochondrial Stress Tests after Repeated Axial Compression
2.7. Statistical Analyses
3. Results
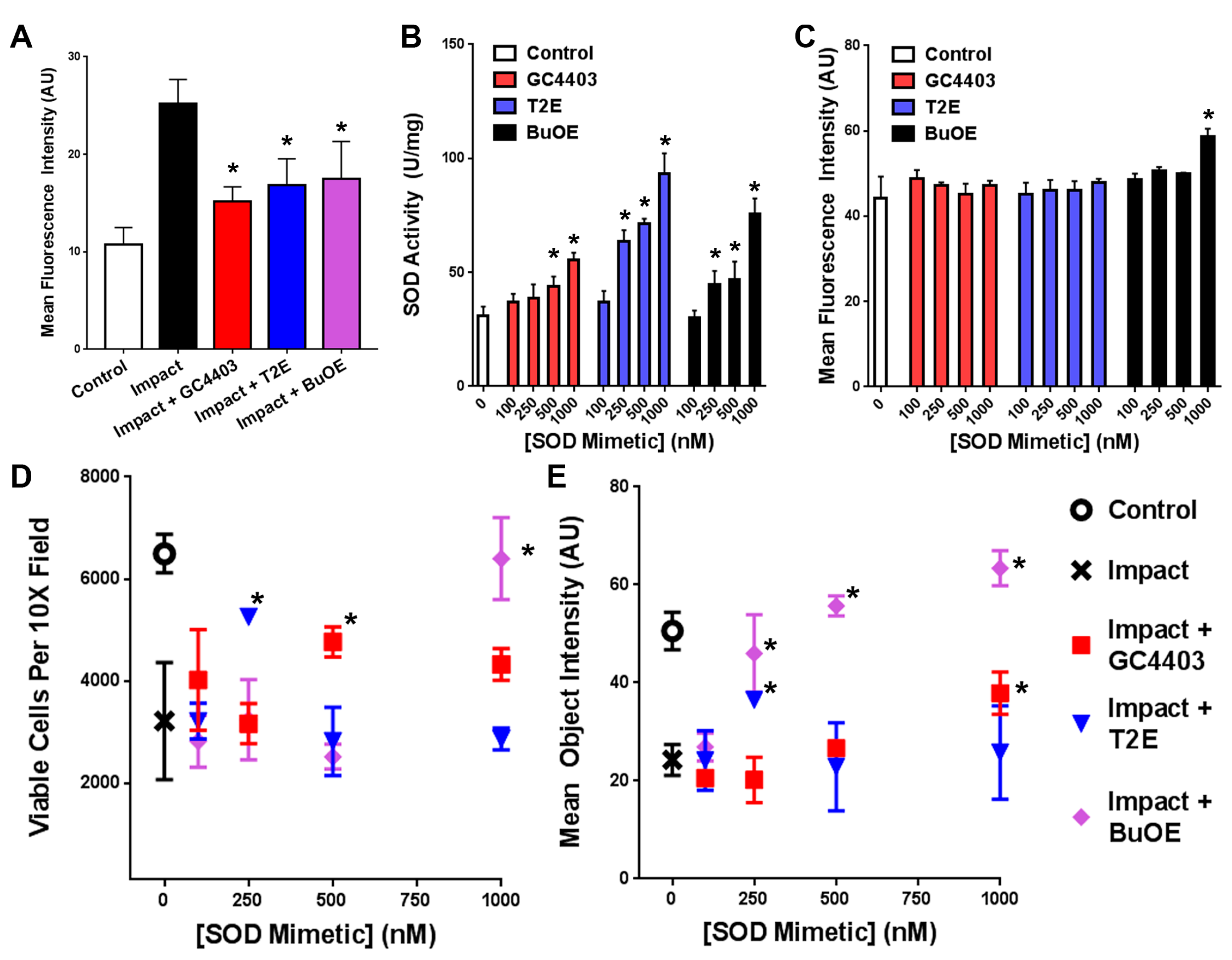
3.1. SOD Mimetics Provide Protection Against Impact-Induced Loss of Mitochondria
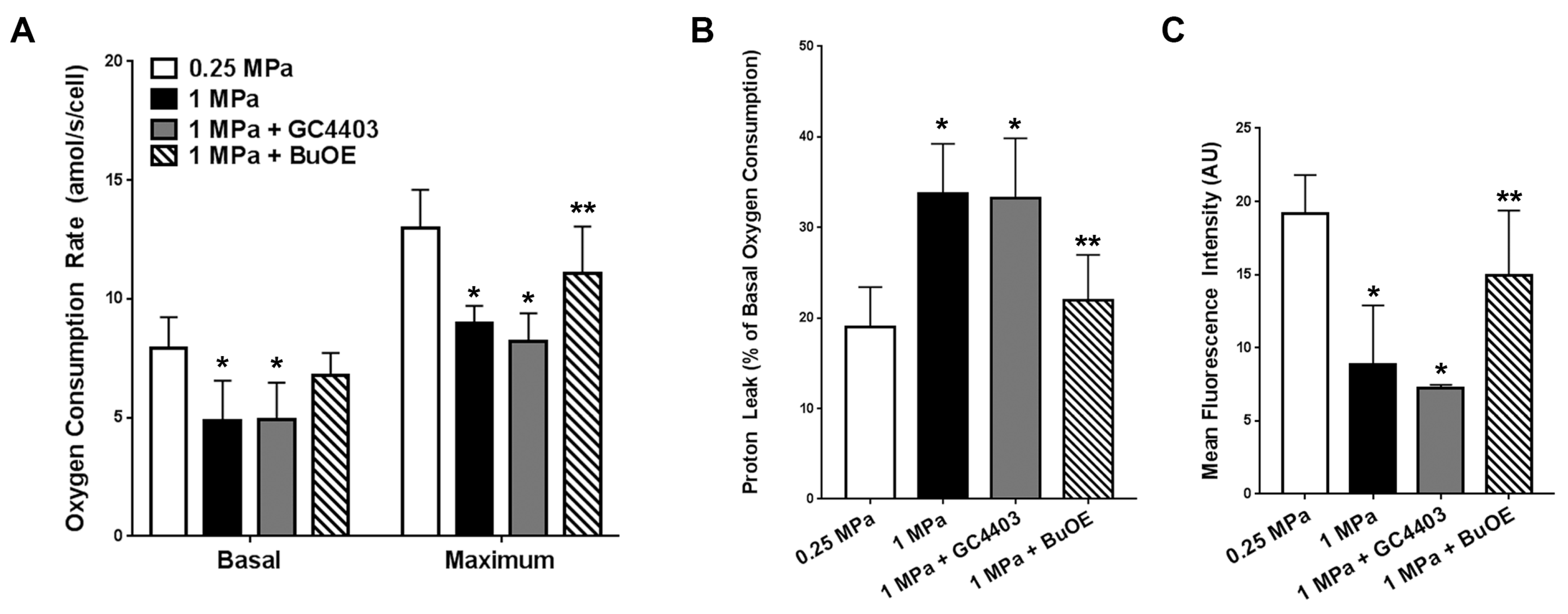
3.2. BuOE Protects against Losses in Mitochondria after Repeated Axial Compression
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brown, T.D.; Johnston, R.C.; Saltzman, C.L.; Marsh, J.L.; Buckwalter, J.A. Posttraumatic Osteoarthritis: A First Estimate of Incidence, Prevalence, and Burden of Disease. J. Orthop. Trauma 2006, 20, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Buckwalter, J.A.; Felson, D.T. Post-Traumatic Arthritis: Definitions and Burden of Disease. In Post-Traumatic Arthritis: Pathogenesis, Diagnosis and Management; Olson, S.A., Guilak, F., Eds.; Springer: New York, NY, USA, 2015; pp. 7–15. [Google Scholar]
- Rivera, J.C.; Wenke, J.C.; Buckwalter, J.A.; Ficke, J.R.; Johnson, A.E. Post-Traumatic Osteoarthritis Caused by Battlefield Injuries is the Primary Source of Disability in Warriors. J. Am. Acad. Orthop. Surg. 2012, 20, S64–S69. [Google Scholar] [CrossRef] [PubMed]
- Honkonen, S.E. Degenerative arthritis after tibial plateau fractures. J. Orthop. Trauma. 1995, 9, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Thomas, M. Tibial plafond fractures—How do these ankles function over time? Fuß SprunggeLenk 2003, 1, 306–307. [Google Scholar] [CrossRef]
- Ossendorf, C.; Kaps, C.; Kreuz, P.C.; Burmester, G.R.; Sittinger, M.; Erggelet, C. Treatment of posttraumatic and focal osteoarthritic cartilage defects of the knee with autologous polymer-based three-dimensional chondrocyte grafts: 2-Year clinical results. Arthritis Res. Ther. 2007, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McKinley, T.O.; Borrelli, J.; D’Lima, D.D.; Furman, B.D.; Giannoudis, P.V. Basic Science of Intra-articular Fractures and Posttraumatic Osteoarthritis. J. Orthop. Trauma 2010, 24, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.L.; Buckwalter, J.A.; Gelberman, R.; Dirschl, D.; Olson, S.; Brown, T.D.; Llinias, A. Articular fractures: Does an anatomic reduction really change the result? J. Bone Joint Surg. Am. 2002, 84, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Matta, J. Fracture of the acetabulum: Accuracy of reduction and clinical results in patients managed operatively within three weeks after the injury. Orthop. Trauma Dir. 2011, 9, 31–36. [Google Scholar] [CrossRef]
- Lattermann, C.; Jacobs, C.A.; Proffitt Bunnell, M.; Huston, L.J.; Gammon, L.G.; Johnson, D.L.; Reinke, E.K.; Huebner, J.L.; Kraus, V.B.; Spindler, K.P. A Multicenter Study of Early Anti-inflammatory Treatment in Patients with Acute Anterior Cruciate Ligament Tear. Am. J. Sports Med. 2017, 45, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.D.; Kilburg, A.T.; Thomas, T.P.; Marsh, J.L. Expedited CT-Based Methods for Evaluating Fracture Severity to Assess Risk of Post-Traumatic Osteoarthritis after Articular Fractures. Iowa Orthop. J. 2016, 36, 46–52. [Google Scholar] [PubMed]
- Thomas, T.P.; Anderson, D.D.; Mosqueda, T.V.; Van Hofwegen, C.J.; Hillis, S.L.; Marsh, J.L.; Brown, T.D. Objective CT-Based Metrics of Articular Fracture Severity to Assess Risk for Posttraumatic Osteoarthritis. J. Orthop. Trauma 2010, 24, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Masrouha, K.Z.; Anderson, D.D.; Thomas, T.P.; Kuhl, L.L.; Brown, T.D.; Marsh, J.L. Acute articular fracture severity and chronic cartilage stress challenge as quantitative risk factors for post-traumatic osteoarthritis: Illustrative cases. Iowa Orthop. J. 2010, 30, 47–54. [Google Scholar] [PubMed]
- Buckwalter, J.A.; Anderson, D.D.; Brown, T.D.; Tochigi, Y.; Martin, J.A. The Roles of Mechanical Stresses in the Pathogenesis of Osteoarthritis: Implications for Treatment of Joint Injuries. Cartilage 2013, 4, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.B.; Urban, J.P. Functional replacement of oxygen by other oxidants in articular cartilage. Arthritis Rheum. 2002, 46, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.B.; Wilkins, R.J.; Razaq, S.; Urban, J.P. The effect of mechanical stress on cartilage energy metabolism. Biorheology 2002, 39, 133–143. [Google Scholar] [PubMed]
- Johnson, K.; Jung, A.; Murphy, A.; Andreyev, A.; Dykens, J.; Terkeltaub, R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000, 43, 1560–1570. [Google Scholar] [CrossRef]
- Terkeltaub, R.; Johnson, K.; Murphy, A.; Ghosh, S. Invited review: The mitochondrion in osteoarthritis. Mitochondrion 2002, 1, 301–319. [Google Scholar] [CrossRef]
- Wolff, K.J.; Ramakrishnan, P.S.; Brouillette, M.J.; Journot, B.J.; McKinley, T.O.; Buckwalter, J.A.; Martin, J.A. Mechanical stress and ATP synthesis are coupled by mitochondrial oxidants in articular cartilage. J. Orthop. Res. 2013, 31, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Brouillette, M.J.; Ramakrishnan, P.S.; Wagner, V.M.; Sauter, E.E.; Journot, B.J.; McKinley, T.O.; Martin, J.A. Strain-dependent oxidant release in articular cartilage originates from mitochondria. Biomech. Model. Mechanobiol. 2013, 13, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, W.; McCabe, D.; Sauter, E.; Reese, E.; Walter, M.; Buckwalter, J.A.; Martin, J.A. Rotenone prevents impact-induced chondrocyte death. J. Orthop. Res. 2010, 28, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.C.; Ramakrishnan, P.S.; Brouillette, M.J.; Martin, J.A. Injurious Loading of Articular Cartilage Compromises Chondrocyte Respiratory Function. Arthritis Rheumatol. 2016, 68, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Nojiri, H.; Ozawa, Y.; Watanabe, K.; Muramatsu, Y.; Kaneko, H.; Morikawa, D.; Kobayashi, K.; Saita, Y.; Sasho, T.; et al. Mechanical overloading causes mitochondrial superoxide and SOD2 imbalance in chondrocytes resulting in cartilage degeneration. Sci. Rep. 2015, 5, 11722. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.L.; Gabrielides, C.; Davidson, R.K.; Swingler, T.E.; Clark, I.M.; Wallis, G.A.; Boot-Handford, R.P.; Kirkwood, T.B.; Taylor, R.W.; Young, D.A. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann. Rheum. Dis. 2010, 69, 1502–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavriilidis, C.; Miwa, S.; von Zglinicki, T.; Taylor, R.W.; Young, D.A. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum. 2013, 65, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Romero, C.; Calamia, V.; Mateos, J.; Carreira, V.; Martinez-Gomariz, M.; Fernandez, M.; Blanco, F.J. Mitochondrial Dysregulation of Osteoarthritic Human Articular Chondrocytes Analyzed by Proteomics: A Decrease in Mitochondrial Superoxide Dismutase Points to a Redox Imbalance. Mol. Cell. Proteom. 2008, 8, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, D.; Wang, Z.Q.; Zweier, J.L.; Samouilov, A.; Macarthur, H.; Misko, T.P.; Currie, M.G.; Cuzzocrea, S.; Sikorski, J.A.; Riley, D.P. A nonpeptidyl mimic of superoxide dismutase with therapeutic activity in rats. Science 1999, 286, 304–306. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, K.M.; Liochev, S.I.; Fridovich, I. Stable Mn(III) porphyrins mimic superoxide dismutase in vitro and substitute for it in vivo. J. Biol. Chem. 1994, 269, 23471–23476. [Google Scholar] [PubMed]
- Rajic, Z.; Tovmasyan, A.; Spasojevic, I.; Sheng, H.; Lu, M.; Li, A.M.; Gralla, E.B.; Warner, D.S.; Benov, L.; Batinic-Haberle, I. A new SOD mimic, Mn(III) ortho N-butoxyethylpyridylporphyrin, combines superb potency and lipophilicity with low toxicity. Free. Radic. Biol. Med. 2012, 52, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Delco, M.L.; Bonnevie, E.D.; Bonassar, L.J.; Fortier, L.A. Mitochondrial dysfunction is an acute response of articular chondrocytes to mechanical injury. J. Orthop. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Spitz, D.R.; Oberley, L.W. An assay for superoxide dismutase activity in mammalian tissue homogenates. Anal. Biochem. 1989, 179, 8–18. [Google Scholar] [CrossRef]
- Mathy-Hartert, M.; Hogge, L.; Sanchez, C.; Deby-Dupont, G.; Crielaard, J.M.; Henrotin, Y. Interleukin-1β and interleukin-6 disturb the antioxidant enzyme system in bovine chondrocytes: A possible explanation for oxidative stress generation. Osteoarthr. Cartil. 2008, 16, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Abusarah, J.; Zaouter, C.; Moldovan, F.; Fernandes, J.C.; Fahmi, H.; Benderdour, M. New Evidence Implicating 4-Hydroxynonenal in the Pathogenesis of Osteoarthritis in vivo. Arthritis Rheumatol. 2014, 66, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Vaillancourt, F.; Fahmi, H.; Shi, Q.; Lavigne, P.; Ranger, P.; Fernandes, J.C.; Benderdour, M. 4-Hydroxynonenal induces apoptosis in human osteoarthritic chondrocytes: The protective role of glutathione-S-transferase. Arthritis Res. Ther. 2008, 10, R107. [Google Scholar] [CrossRef] [PubMed]
- Regan, E.; Flannelly, J.; Bowler, R.; Tran, K.; Nicks, M.; Carbone, B.D.; Glueck, D.; Heijnen, H.; Mason, R.; Crapo, J. Extracellular superoxide dismutase and oxidant damage in osteoarthritis. Arthritis Rheum. 2005, 52, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.M.; Sherk, V.D.; Carpenter, R.D.; Weaver, M.; Crapo, S.; Gally, F.; Chatham, L.S.; Goldstrohm, D.A.; Crapo, J.D.; Kohrt, W.M.; et al. The beneficial effects of exercise on cartilage are lost in mice with reduced levels of ECSOD in tissues. J. Appl. Physiol. 2015, 118, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Gammer, W.; Brobäck, L.-G. Clinical Comparison of Orgotein and Methylpredisolone acetate in the Treatment of Osteoarthrosis of the Knee Joint. Scand. J. Rheumatol. 1984, 13, 108–112. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coleman, M.C.; Brouillette, M.J.; Andresen, N.S.; Oberley-Deegan, R.E.; Martin, J.M. Differential Effects of Superoxide Dismutase Mimetics after Mechanical Overload of Articular Cartilage. Antioxidants 2017, 6, 98. https://doi.org/10.3390/antiox6040098
Coleman MC, Brouillette MJ, Andresen NS, Oberley-Deegan RE, Martin JM. Differential Effects of Superoxide Dismutase Mimetics after Mechanical Overload of Articular Cartilage. Antioxidants. 2017; 6(4):98. https://doi.org/10.3390/antiox6040098
Chicago/Turabian StyleColeman, Mitchell C., Marc J. Brouillette, Nicholas S. Andresen, Rebecca E. Oberley-Deegan, and James M. Martin. 2017. "Differential Effects of Superoxide Dismutase Mimetics after Mechanical Overload of Articular Cartilage" Antioxidants 6, no. 4: 98. https://doi.org/10.3390/antiox6040098
APA StyleColeman, M. C., Brouillette, M. J., Andresen, N. S., Oberley-Deegan, R. E., & Martin, J. M. (2017). Differential Effects of Superoxide Dismutase Mimetics after Mechanical Overload of Articular Cartilage. Antioxidants, 6(4), 98. https://doi.org/10.3390/antiox6040098