
ROS Cocktails as an Adjuvant for Personalized Antitumor Vaccination?

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tumor Immune Evasion and Vaccination
3. Reactive Oxygen and Nitrogen Species
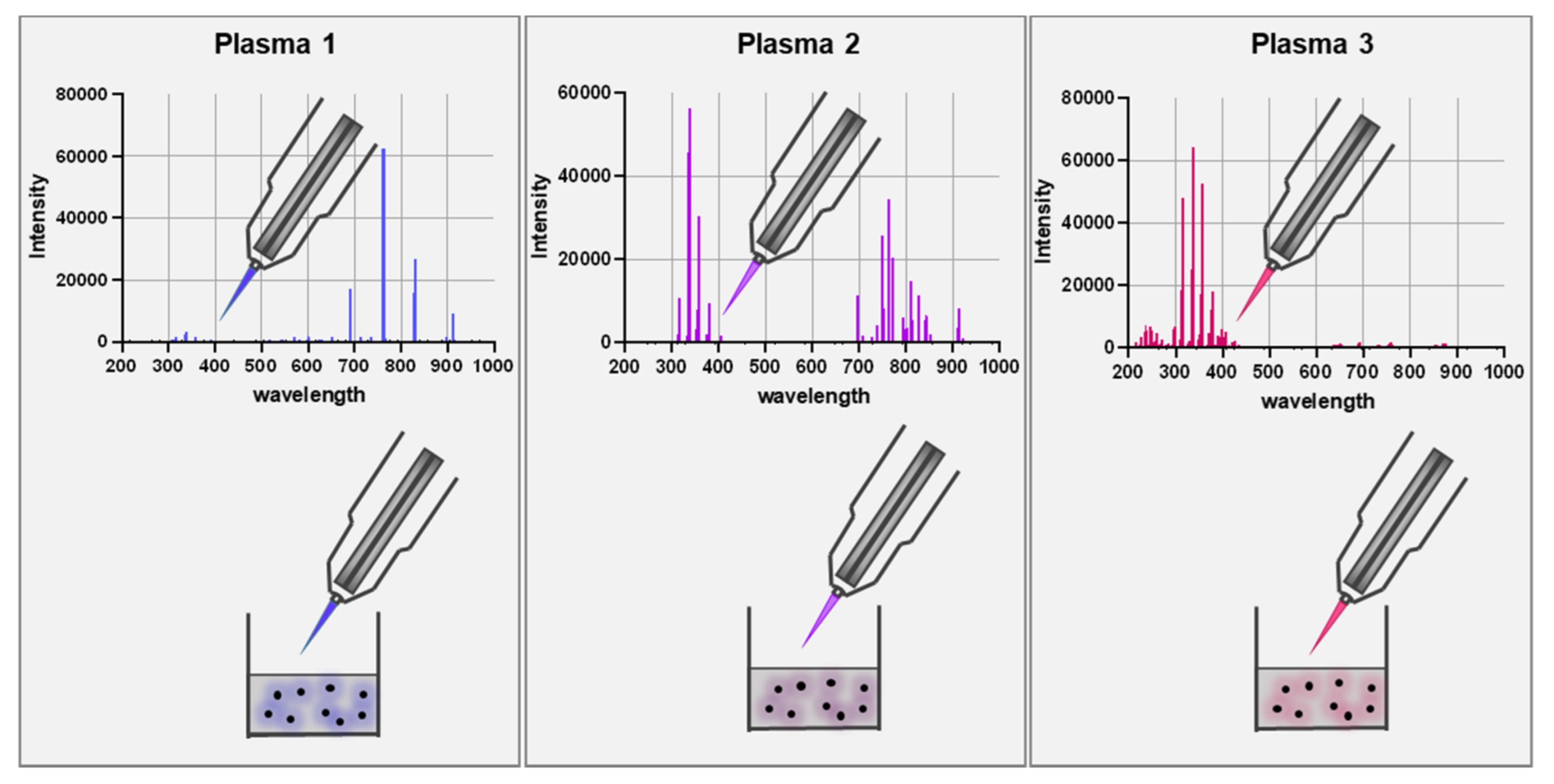
4. Gas Plasma Technology as a Significant Innovation in Generating Multi-ROS Cocktails
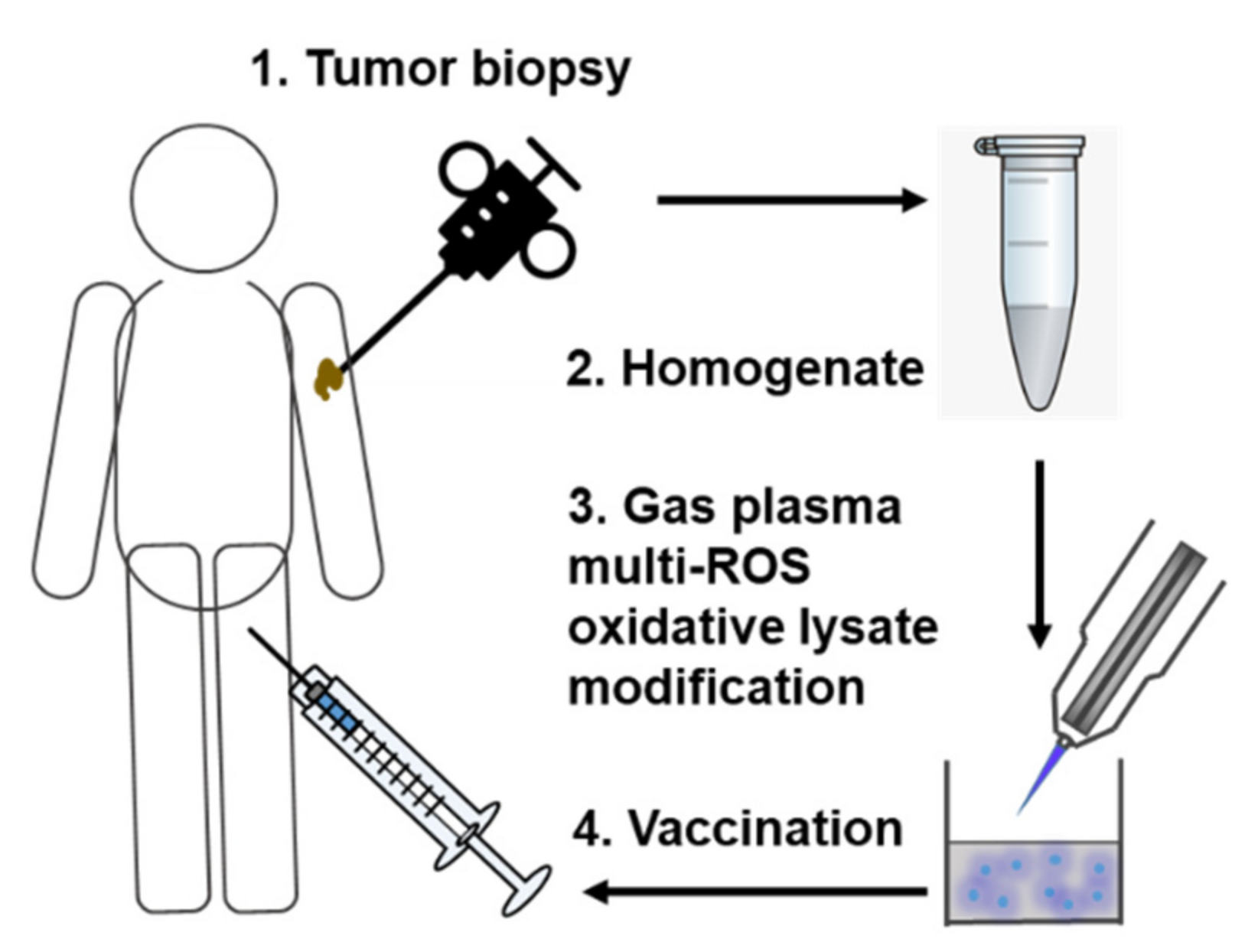
5. Proof-of-Concept Study Using Multi-ROS Cocktails to Provide Vaccine Tumor Control
6. Concept and Challenges of Multi-ROS-Modified Autologous Tumor Vaccines
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [Green Version]
- Ghetie, M.-A.; Ghetie, V.; Vitetta, E.S. Section Review Biologicals & Immunologicals: The use of immunoconjugates in cancer therapy. Expert Opin. Investig. Drugs 2008, 5, 309–321. [Google Scholar] [CrossRef]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Piessens, W.F. Evidence for human cancer immunity. A review. Cancer 1970, 26, 1212–1220. [Google Scholar] [CrossRef]
- Finney, J.W.; Byers, E.H.; Wilson, R.H. Studies in Tumor Auto-Immunity. Cancer Res. 1960, 20, 351–356. [Google Scholar]
- Azoury, S.C.; Straughan, D.M.; Shukla, V. Immune Checkpoint Inhibitors for Cancer Therapy: Clinical Efficacy and Safety. Curr. Cancer Drug Targets 2015, 15, 452–462. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Wang, T.; Wang, D.; Yu, H.; Feng, B.; Zhou, F.; Zhang, H.; Zhou, L.; Jiao, S.; Li, Y. A cancer vaccine-mediated postoperative immunotherapy for recurrent and metastatic tumors. Nat. Commun. 2018, 9, 1532. [Google Scholar] [CrossRef] [Green Version]
- Kotsakis, A.; Vetsika, E.K.; Christou, S.; Hatzidaki, D.; Vardakis, N.; Aggouraki, D.; Konsolakis, G.; Georgoulias, V.; Christophyllakis, C.; Cordopatis, P.; et al. Clinical outcome of patients with various advanced cancer types vaccinated with an optimized cryptic human telomerase reverse transcriptase (TERT) peptide: Results of an expanded phase II study. Ann. Oncol. 2012, 23, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Goldinger, S.M.; Dummer, R.; Baumgaertner, P.; Mihic-Probst, D.; Schwarz, K.; Hammann-Haenni, A.; Willers, J.; Geldhof, C.; Prior, J.O.; Kundig, T.M.; et al. Nano-particle vaccination combined with TLR-7 and -9 ligands triggers memory and effector CD8(+) T-cell responses in melanoma patients. Eur. J. Immunol. 2012, 42, 3049–3061. [Google Scholar] [CrossRef] [Green Version]
- Topfer, K.; Kempe, S.; Muller, N.; Schmitz, M.; Bachmann, M.; Cartellieri, M.; Schackert, G.; Temme, A. Tumor evasion from T cell surveillance. J. Biomed. Biotechnol. 2011, 2011, 918471. [Google Scholar] [CrossRef] [Green Version]
- Poschke, I.; Mougiakakos, D.; Kiessling, R. Camouflage and sabotage: Tumor escape from the immune system. Cancer Immunol. Immunother. 2011, 60, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Sosman, J.A.; Zhang, B.; Wu, J.D.; Miller, S.D.; Meeks, J.J.; et al. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front. Immunol. 2020, 11, 1185. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Elkord, E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin. Cancer Biol. 2020, 65, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Dalet, A.; Stroobant, V.; Vigneron, N.; Van den Eynde, B.J. Differences in the production of spliced antigenic peptides by the standard proteasome and the immunoproteasome. Eur. J. Immunol. 2011, 41, 39–46. [Google Scholar] [CrossRef]
- Van den Eynde, B.T.J.; Morel, S. Differential processing of class-I-restricted epitopes by the standard proteasome and the immunoproteasome. Curr. Opin. Immunol. 2001, 13, 147–153. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Kloetzel, P.M. The proteasome and MHC class I antigen processing. Biochim. Biophys. Acta 2004, 1695, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.Z.; Zhao, X.; Song, X.R. Ex vivo pulsed dendritic cell vaccination against cancer. Acta Pharmacol. Sin. 2020, 41, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Farkas, A.; Conrad, C. Dendritic-cell-based therapeutic vaccination against cancer. Curr. Opin. Immunol. 2005, 17, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, T.J.; Olson, K.B.; Laffin, R.; Horton, J.; Sullivan, J. Treatment of advanced cancer with active immunization. Cancer 1969, 24, 932–937. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Jahan, T.; Ross, H.; Sterman, D.; Richards, D.; Fox, B.; Jablons, D.; Aimi, J.; Lin, A.; Hege, K. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther. 2006, 13, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Saini, R.; Lee, N.V.; Liu, K.Y.; Poh, C.F. Prospects in the Application of Photodynamic Therapy in Oral Cancer and Premalignant Lesions. Cancers 2016, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Schwaab, T.; Tretter, C.; Gibson, J.J.; Cole, B.F.; Schned, A.R.; Harris, R.; Wallen, E.M.; Fisher, J.L.; Waugh, M.G.; Truman, D.; et al. Immunological effects of granulocyte-macrophage colony-stimulating factor and autologous tumor vaccine in patients with renal cell carcinoma. J. Urol. 2004, 171, 1036–1042. [Google Scholar] [CrossRef]
- Olin, M.R.; Pluhar, G.E.; Andersen, B.M.; Shaver, R.; Waldron, N.N.; Moertel, C.L. Victory and defeat in the induction of a therapeutic response through vaccine therapy for human and canine brain tumors: A review of the state of the art. Crit. Rev. Immunol. 2014, 34, 399–432. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ma, B.; Wang, W. Peptide-Based Nanomaterials for Tumor Immunotherapy. Molecules 2020, 26, 132. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins—Molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 2013, 18, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brustle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017, 46, 675–689. [Google Scholar] [CrossRef] [Green Version]
- Rashida Gnanaprakasam, J.N.; Wu, R.; Wang, R. Metabolic Reprogramming in Modulating T Cell Reactive Oxygen Species Generation and Antioxidant Capacity. Front. Immunol. 2018, 9, 1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [Green Version]
- Stanley, C.P.; Maghzal, G.J.; Ayer, A.; Talib, J.; Giltrap, A.M.; Shengule, S.; Wolhuter, K.; Wang, Y.; Chadha, P.; Suarna, C.; et al. Singlet molecular oxygen regulates vascular tone and blood pressure in inflammation. Nature 2019, 566, 548–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Hirshberg, J.; Lyle, D.; Freij, J.B.; Caturegli, P. Reactive oxygen species in organ-specific autoimmunity. Auto Immun Highlights 2016, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, H.R. Is the generation of neo-antigenic determinants by free radicals central to the development of autoimmune rheumatoid disease? Autoimmun. Rev. 2008, 7, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Arif, Z.; Neelofar, K.; Tarannum, A.; Arfat, M.Y.; Ahmad, S.; Zaman, A.; Khan, M.A.; Badar, A.; Islam, S.N.; Iqubal, M.A. SLE autoantibodies are well recognized by peroxynitrite-modified-HSA: Its implications in the pathogenesis of SLE. Int. J. Biol. Macromol. 2018, 106, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Kurien, B.T.; Scofield, R.H. Autoimmunity and oxidatively modified autoantigens. Autoimmun. Rev. 2008, 7, 567–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nybo, T.; Dieterich, S.; Gamon, L.F.; Chuang, C.Y.; Hammer, A.; Hoefler, G.; Malle, E.; Rogowska-Wrzesinska, A.; Davies, M.J. Chlorination and oxidation of the extracellular matrix protein laminin and basement membrane extracts by hypochlorous acid and myeloperoxidase. Redox Biol. 2019, 20, 496–513. [Google Scholar] [CrossRef]
- Strollo, R.; Vinci, C.; Arshad, M.H.; Perrett, D.; Tiberti, C.; Chiarelli, F.; Napoli, N.; Pozzilli, P.; Nissim, A. Antibodies to post-translationally modified insulin in type 1 diabetes. Diabetologia 2015, 58, 2851–2860. [Google Scholar] [CrossRef] [Green Version]
- Mannering, S.I.; Di Carluccio, A.R.; Elso, C.M. Neoepitopes: A new take on beta cell autoimmunity in type 1 diabetes. Diabetologia 2019, 62, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.L.; Doyle, H.A.; Clarke, S.G.; Herold, K.C.; Mamula, M.J. Oxidative Modifications in Tissue Pathology and Autoimmune Disease. Antioxid. Redox Signal. 2018, 29, 1415–1431. [Google Scholar] [CrossRef]
- Carta, S.; Castellani, P.; Delfino, L.; Tassi, S.; Vene, R.; Rubartelli, A. DAMPs and inflammatory processes: The role of redox in the different outcomes. J. Leukoc. Biol. 2009, 86, 549–555. [Google Scholar] [CrossRef]
- Winter, J.; Brandenburg, R.; Weltmann, K.D. Atmospheric pressure plasma jets: An overview of devices and new directions. Plasma Sources Sci. Technol. 2015, 24, 064001. [Google Scholar] [CrossRef]
- Privat-Maldonado, A.; Schmidt, A.; Lin, A.; Weltmann, K.D.; Wende, K.; Bogaerts, A.; Bekeschus, S. ROS from Physical Plasmas: Redox Chemistry for Biomedical Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 9062098. [Google Scholar] [CrossRef] [Green Version]
- Graves, D.B. Mechanisms of Plasma Medicine: Coupling Plasma Physics, Biochemistry, and Biology. IEEE Trans. Radiat. Plasma Med. Sci. 2017, 1, 281–292. [Google Scholar] [CrossRef]
- Von Woedtke, T.; Schmidt, A.; Bekeschus, S.; Wende, K.; Weltmann, K.D. Plasma Medicine: A Field of Applied Redox Biology. In Vivo 2019, 33, 1011–1026. [Google Scholar] [CrossRef] [Green Version]
- Wende, K.; von Woedtke, T.; Weltmann, K.D.; Bekeschus, S. Chemistry and biochemistry of cold physical plasma derived reactive species in liquids. Biol. Chem. 2018, 400, 19–38. [Google Scholar] [CrossRef]
- Schmidt-Bleker, A.; Winter, J.; Iseni, S.; Dunnbier, M.; Weltmann, K.D.; Reuter, S. Reactive species output of a plasma jet with a shielding gas device-combination of FTIR absorption spectroscopy and gas phase modelling. J. Phys. D Appl. Phys. 2014, 47, 145201. [Google Scholar] [CrossRef]
- Jablonowski, H.; Santos Sousa, J.; Weltmann, K.D.; Wende, K.; Reuter, S. Quantification of the ozone and singlet delta oxygen produced in gas and liquid phases by a non-thermal atmospheric plasma with relevance for medical treatment. Sci. Rep. 2018, 8, 12195. [Google Scholar] [CrossRef]
- Jablonowski, H.; Schmidt-Bleker, A.; Weltmann, K.D.; von Woedtke, T.; Wende, K. Non-touching plasma-liquid interaction—Where is aqueous nitric oxide generated? Phys. Chem. Chem. Phys. 2018, 20, 25387–25398. [Google Scholar] [CrossRef] [Green Version]
- Lackmann, J.W.; Wende, K.; Verlackt, C.; Golda, J.; Volzke, J.; Kogelheide, F.; Held, J.; Bekeschus, S.; Bogaerts, A.; Schulz-von der Gathen, V.; et al. Chemical fingerprints of cold physical plasmas—An experimental and computational study using cysteine as tracer compound. Sci. Rep. 2018, 8, 7736. [Google Scholar] [CrossRef]
- Wenske, S.; Lackmann, J.-W.; Bekeschus, S.; Weltmann, K.-D.; von Woedtke, T.; Wende, K. Nonenzymatic post-translational modifications in peptides by cold plasma-derived reactive oxygen and nitrogen species. Biointerphases 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Yusupov, M.; Lackmann, J.-W.; Razzokov, J.; Kumar, S.; Stapelmann, K.; Bogaerts, A. Impact of plasma oxidation on structural features of human epidermal growth factor. Plasma Process. Polym. 2018, 15. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, J.; Shen, J.; Lan, Y.; Ding, L.; Qian, S.; Cheng, C.; Xia, W.; Chu, P.K. Comparison of the Effects Induced by Plasma Generated Reactive Species and H2O2 on Lactate Dehydrogenase (LDH) Enzyme. IEEE Trans. Plasma Sci. 2018, 46, 2742–2752. [Google Scholar] [CrossRef]
- Krewing, M.; Stepanek, J.J.; Cremers, C.; Lackmann, J.W.; Schubert, B.; Muller, A.; Awakowicz, P.; Leichert, L.I.O.; Jakob, U.; Bandow, J.E. The molecular chaperone Hsp33 is activated by atmospheric-pressure plasma protecting proteins from aggregation. J. R. Soc. Interface 2019, 16, 20180966. [Google Scholar] [CrossRef]
- Krewing, M.; Jung, C.T.K.; Dobbelstein, E.; Schubert, B.; Jacob, T.; Bandow, J.E. Dielectric barrier discharge plasma treatment affects stability, metal ion coordination, and enzyme activity of bacterial superoxide dismutases. Plasma Process. Polym. 2020, 17. [Google Scholar] [CrossRef]
- Clemen, R.; Freund, E.; Mrochen, D.; Miebach, L.; Schmidt, A.; Rauch, B.H.; Lackmann, J.W.; Martens, U.; Wende, K.; Lalk, M.; et al. Gas Plasma Technology Augments Ovalbumin Immunogenicity and OT-II T Cell Activation Conferring Tumor Protection in Mice. Adv. Sci. 2021. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Khalili, M.; Daniels, L.; Lin, A.; Krebs, F.C.; Snook, A.E.; Bekeschus, S.; Bowne, W.B.; Miller, V. Non-Thermal Plasma-Induced Immunogenic Cell Death in Cancer: A Topical Review. J. Phys. D Appl. Phys. 2019, 52. [Google Scholar] [CrossRef]
- De Backer, J.; Razzokov, J.; Hammerschmid, D.; Mensch, C.; Hafideddine, Z.; Kumar, N.; van Raemdonck, G.; Yusupov, M.; Van Doorslaer, S.; Johannessen, C.; et al. The effect of reactive oxygen and nitrogen species on the structure of cytoglobin: A potential tumor suppressor. Redox Biol. 2018, 19, 1–10. [Google Scholar] [CrossRef]
- Bekeschus, S.; Ressel, V.; Freund, E.; Gelbrich, N.; Mustea, A.; Stope, M.B. Gas Plasma-Treated Prostate Cancer Cells Augment Myeloid Cell Activity and Cytotoxicity. Antioxidants 2020, 9, 323. [Google Scholar] [CrossRef]
- Poulsen, T.B.G.; Damgaard, D.; Jorgensen, M.M.; Senolt, L.; Blackburn, J.M.; Nielsen, C.H.; Stensballe, A. Identification of Novel Native Autoantigens in Rheumatoid Arthritis. Biomedicines 2020, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Ge, C.; Lonnblom, E.; Lin, X.; Feng, H.; Xiao, L.; Bai, J.; Ayoglu, B.; Nilsson, P.; Nandakumar, K.S.; et al. The autoantibody response to cyclic citrullinated collagen type II peptides in rheumatoid arthritis. Rheumatology 2019, 58, 1623–1633. [Google Scholar] [CrossRef]
- Sidney, J.; Vela, J.L.; Friedrich, D.; Kolla, R.; von Herrath, M.; Wesley, J.D.; Sette, A. Low HLA binding of diabetes-associated CD8+ T-cell epitopes is increased by post translational modifications. BMC Immunol. 2018, 19, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Vomund, A.N.; Peterson, O.J.; Chervonsky, A.V.; Lichti, C.F.; Unanue, E.R. The MHC-II peptidome of pancreatic islets identifies key features of autoimmune peptides. Nat. Immunol. 2020, 21, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Hultqvist, M.; Olofsson, P.; Gelderman, K.A.; Holmberg, J.; Holmdahl, R. A new arthritis therapy with oxidative burst inducers. PLoS Med. 2006, 3, e348. [Google Scholar] [CrossRef] [Green Version]
- Hultqvist, M.; Backlund, J.; Bauer, K.; Gelderman, K.A.; Holmdahl, R. Lack of reactive oxygen species breaks T cell tolerance to collagen type II and allows development of arthritis in mice. J. Immunol. 2007, 179, 1431–1437. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.L.; Coukos, G.; Kandalaft, L.E. Whole Tumor Antigen Vaccines: Where Are We? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.L.; Hagemann, A.R.; Leskowitz, R.; Mick, R.; Garrabrant, T.; Czerniecki, B.J.; Kandalaft, L.E.; Powell, D.J., Jr.; Coukos, G. Day-4 myeloid dendritic cells pulsed with whole tumor lysate are highly immunogenic and elicit potent anti-tumor responses. PLoS ONE 2011, 6, e28732. [Google Scholar] [CrossRef]
- Chiang, C.L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Smith, L.; Nisenbaum, H.L.; et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: From bench to bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef] [Green Version]
- Kandalaft, L.E.; Chiang, C.L.; Tanyi, J.; Motz, G.; Balint, K.; Mick, R.; Coukos, G. A Phase I vaccine trial using dendritic cells pulsed with autologous oxidized lysate for recurrent ovarian cancer. J. Transl. Med. 2013, 11, 149. [Google Scholar] [CrossRef] [Green Version]
- Martin Lluesma, S.; Wolfer, A.; Harari, A.; Kandalaft, L.E. Cancer Vaccines in Ovarian Cancer: How Can We Improve? Biomedicines 2016, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, A.; Graciotti, M.; Kandalaft, L.E.; Kandalaft, L. A cancer vaccine with dendritic cells differentiated with GM-CSF and IFNalpha and pulsed with a squaric acid treated cell lysate improves T cell priming and tumor growth control in a mouse model. Bioimpacts 2018, 8, 211–221. [Google Scholar] [CrossRef]
- Ophir, E.; Bobisse, S.; Coukos, G.; Harari, A.; Kandalaft, L.E. Personalized approaches to active immunotherapy in cancer. Biochim. Biophys. Acta 2016, 1865, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Powell, D.J., Jr.; Singh, N.; Coukos, G. Immunotherapy for ovarian cancer: What’s next? J. Clin. Oncol. 2011, 29, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Singh, N.; Liao, J.B.; Facciabene, A.; Berek, J.S.; Powell, D.J., Jr.; Coukos, G. The emergence of immunomodulation: Combinatorial immunochemotherapy opportunities for the next decade. Gynecol. Oncol. 2010, 116, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.G.; Xiang, B.; Merlino, D.J.; Baybutt, T.R.; Sahu, J.; Fridman, A.; Snook, A.E.; Miller, V. Non-thermal plasma induces immunogenic cell death in vivo in murine CT26 colorectal tumors. Oncoimmunology 2018, 7, e1484978. [Google Scholar] [CrossRef] [Green Version]
- Bekeschus, S.; Clemen, R.; Niessner, F.; Sagwal, S.K.; Freund, E.; Schmidt, A. Medical Gas Plasma Jet Technology Targets Murine Melanoma in an Immunogenic Fashion. Adv. Sci. 2020, 7, 1903438. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Gorbanev, Y.; De Backer, J.; Van Loenhout, J.; Van Boxem, W.; Lemiere, F.; Cos, P.; Dewilde, S.; Smits, E.; Bogaerts, A. Non-Thermal Plasma as a Unique Delivery System of Short-Lived Reactive Oxygen and Nitrogen Species for Immunogenic Cell Death in Melanoma Cells. Adv. Sci. 2019, 6, 1802062. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, K.; Sho, M.; Wakatsuki, K.; Takayama, T.; Matsumoto, S.; Nakamura, S.; Akahori, T.; Tanaka, T.; Migita, K.; Ito, M.; et al. Prognostic importance of tumour-infiltrating memory T cells in oesophageal squamous cell carcinoma. Clin. Exp. Immunol. 2012, 168, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Koelzer, V.H.; Lugli, A.; Dawson, H.; Hadrich, M.; Berger, M.D.; Borner, M.; Mallaev, M.; Galvan, J.A.; Amsler, J.; Schnuriger, B.; et al. CD8/CD45RO T-cell infiltration in endoscopic biopsies of colorectal cancer predicts nodal metastasis and survival. J. Transl. Med. 2014, 12, 81. [Google Scholar] [CrossRef] [Green Version]
- Lamberti, M.J.; Nigro, A.; Mentucci, F.M.; Rumie Vittar, N.B.; Casolaro, V.; Dal Col, J. Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity. Pharmaceutics 2020, 12, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef] [PubMed]
- Vassilaros, S.; Tsibanis, A.; Tsikkinis, A.; Pietersz, G.A.; McKenzie, I.F.; Apostolopoulos, V. Up to 15-year clinical follow-up of a pilot Phase III immunotherapy study in stage II breast cancer patients using oxidized mannan-MUC1. Immunotherapy 2013, 5, 1177–1182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-X.; Sun, X.-M.; Jia, Y.-B.; Liu, X.-G.; Dong, M.; Xu, Z.P.; Liu, R.-T. Nanovaccine’s rapid induction of anti-tumor immunity significantly improves malignant cancer immunotherapy. Nano Today 2020, 35. [Google Scholar] [CrossRef]
- Stone, J.D.; Chervin, A.S.; Kranz, D.M. T-cell receptor binding affinities and kinetics: Impact on T-cell activity and specificity. Immunology 2009, 126, 165–176. [Google Scholar] [CrossRef]
- Baumgartner, C.K.; Yagita, H.; Malherbe, L.P. A TCR affinity threshold regulates memory CD4 T cell differentiation following vaccination. J. Immunol. 2012, 189, 2309–2317. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clemen, R.; Bekeschus, S. ROS Cocktails as an Adjuvant for Personalized Antitumor Vaccination? Vaccines 2021, 9, 527. https://doi.org/10.3390/vaccines9050527
Clemen R, Bekeschus S. ROS Cocktails as an Adjuvant for Personalized Antitumor Vaccination? Vaccines. 2021; 9(5):527. https://doi.org/10.3390/vaccines9050527
Chicago/Turabian StyleClemen, Ramona, and Sander Bekeschus. 2021. "ROS Cocktails as an Adjuvant for Personalized Antitumor Vaccination?" Vaccines 9, no. 5: 527. https://doi.org/10.3390/vaccines9050527
APA StyleClemen, R., & Bekeschus, S. (2021). ROS Cocktails as an Adjuvant for Personalized Antitumor Vaccination? Vaccines, 9(5), 527. https://doi.org/10.3390/vaccines9050527