
A Combined Ultrafiltration/Diafiltration Process for the Purification of Oncolytic Measles Virus



Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Production and Pretreatment
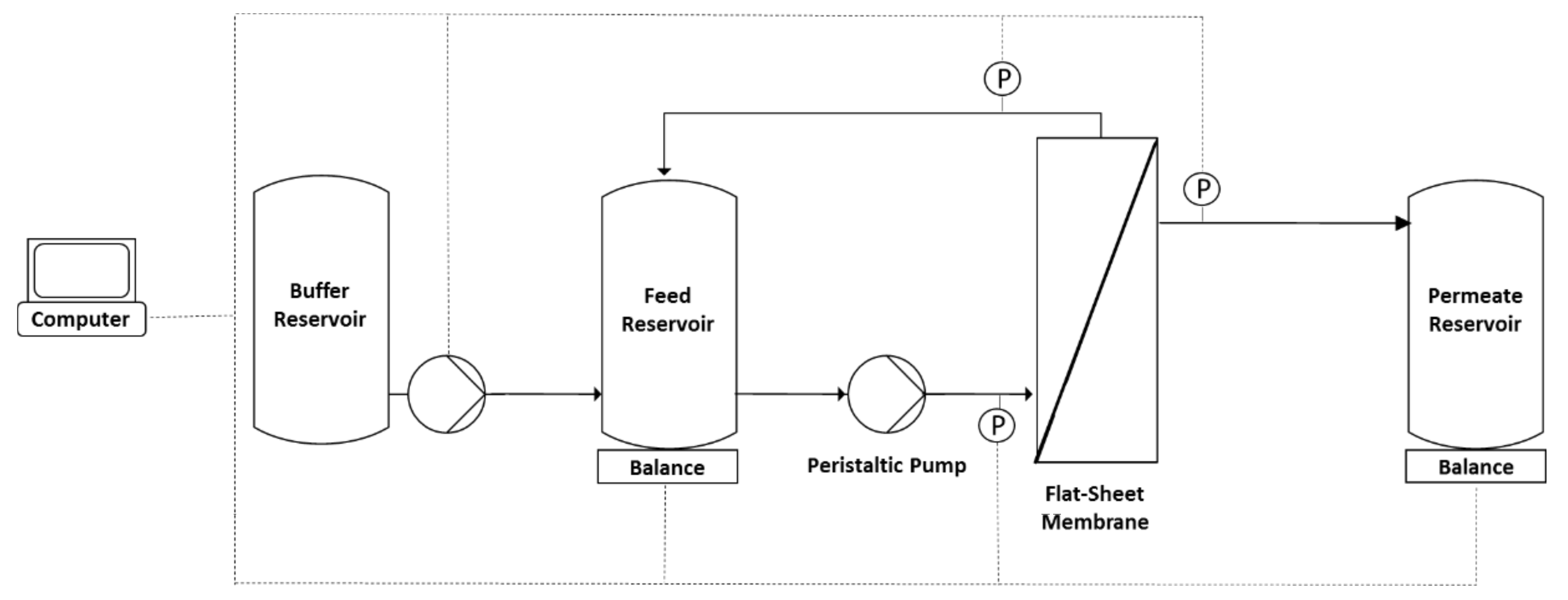
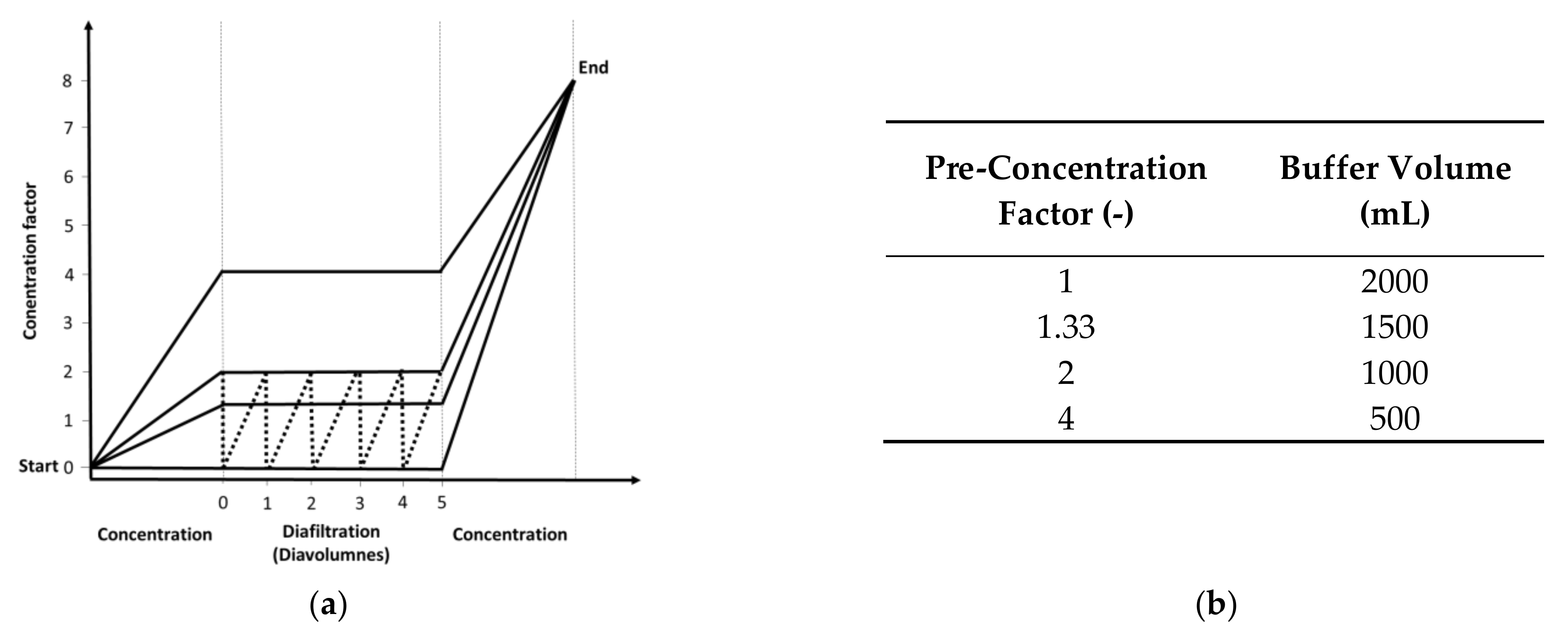
2.2. Diafiltration Experiments
2.3. Assays
2.3.1. MV Infectivity Assay
2.3.2. Quantification of Total Viral RNA
2.3.3. Quantification of Total Protein
2.3.4. Quantification of Host Cell DNA
2.4. Calculations
3. Results
3.1. Characterization of MV Suspensions
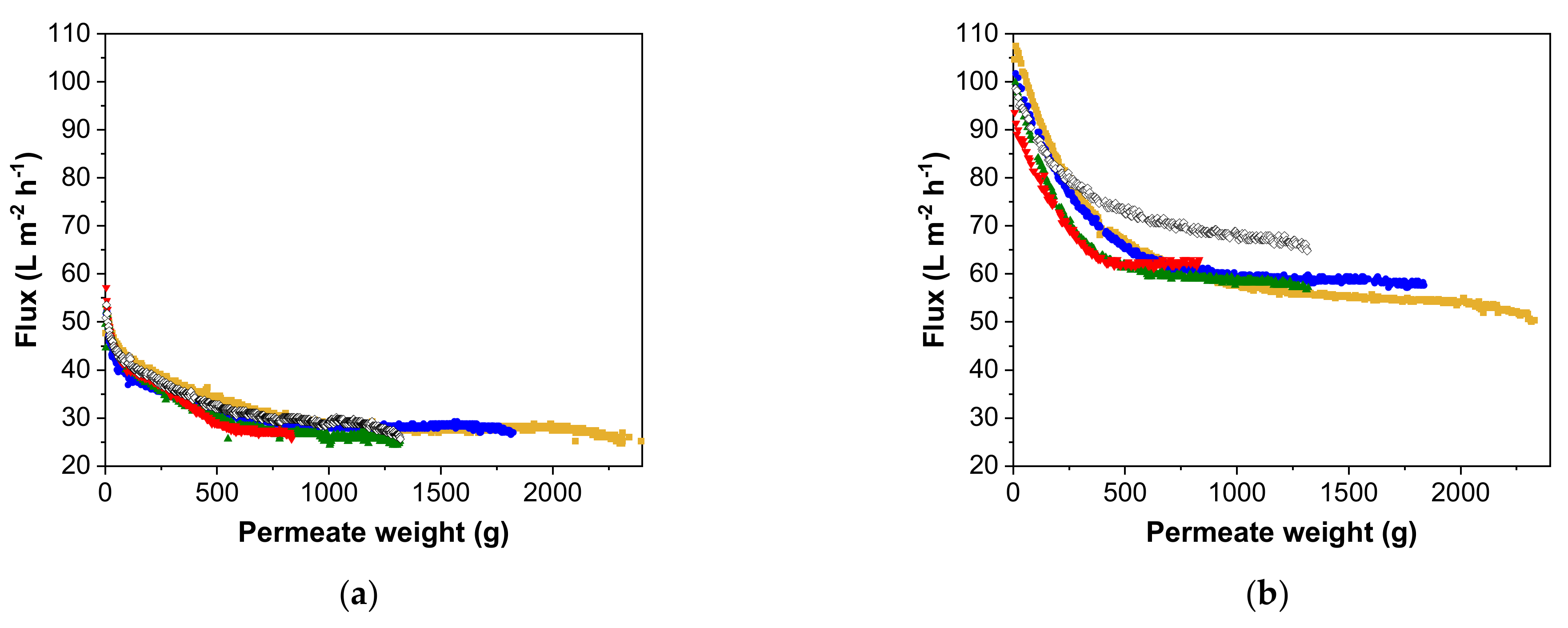
3.2. Flux Behavior Based on Different Pre-Concentration Factors
3.3. Concentration Mode
3.3.1. Infectious Virus Recovery
3.3.2. Retention Factors for Viral RNA and Impurities (hcDNA and Total Proteins)
3.4. Diafiltration Mode
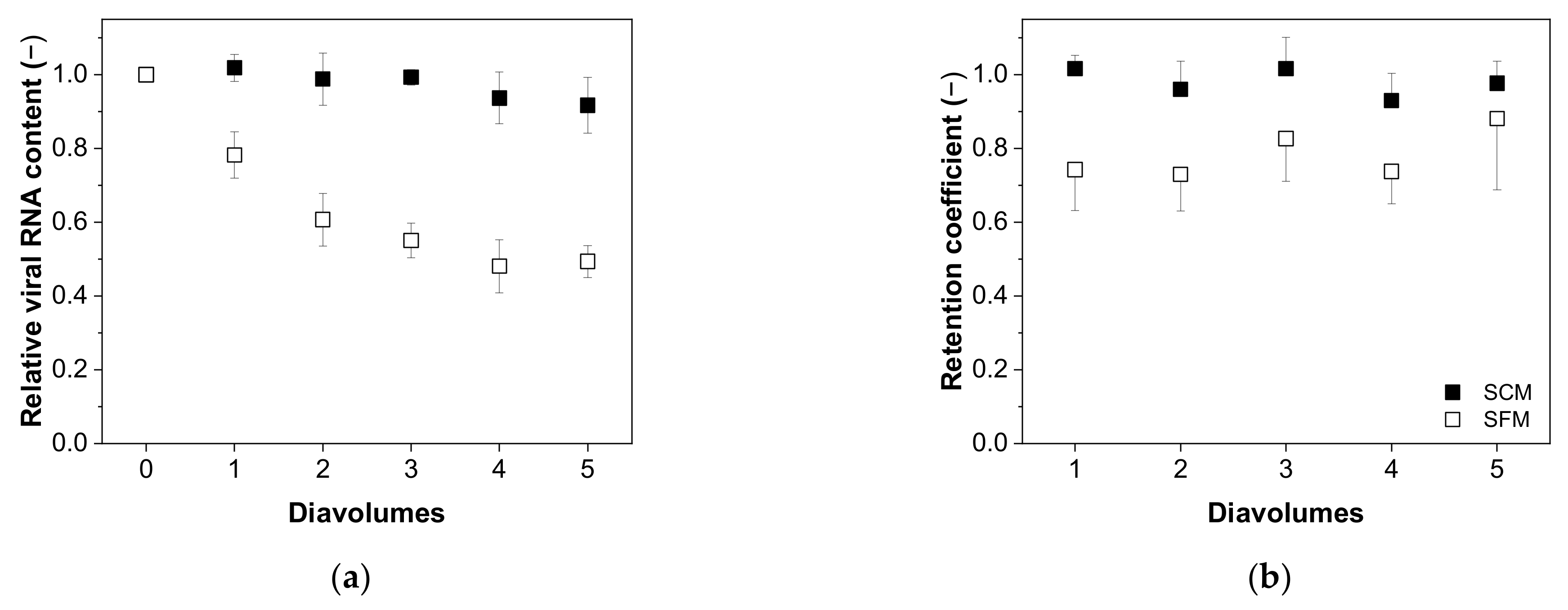
3.4.1. Recovery of Total and Infectious Virus Particles
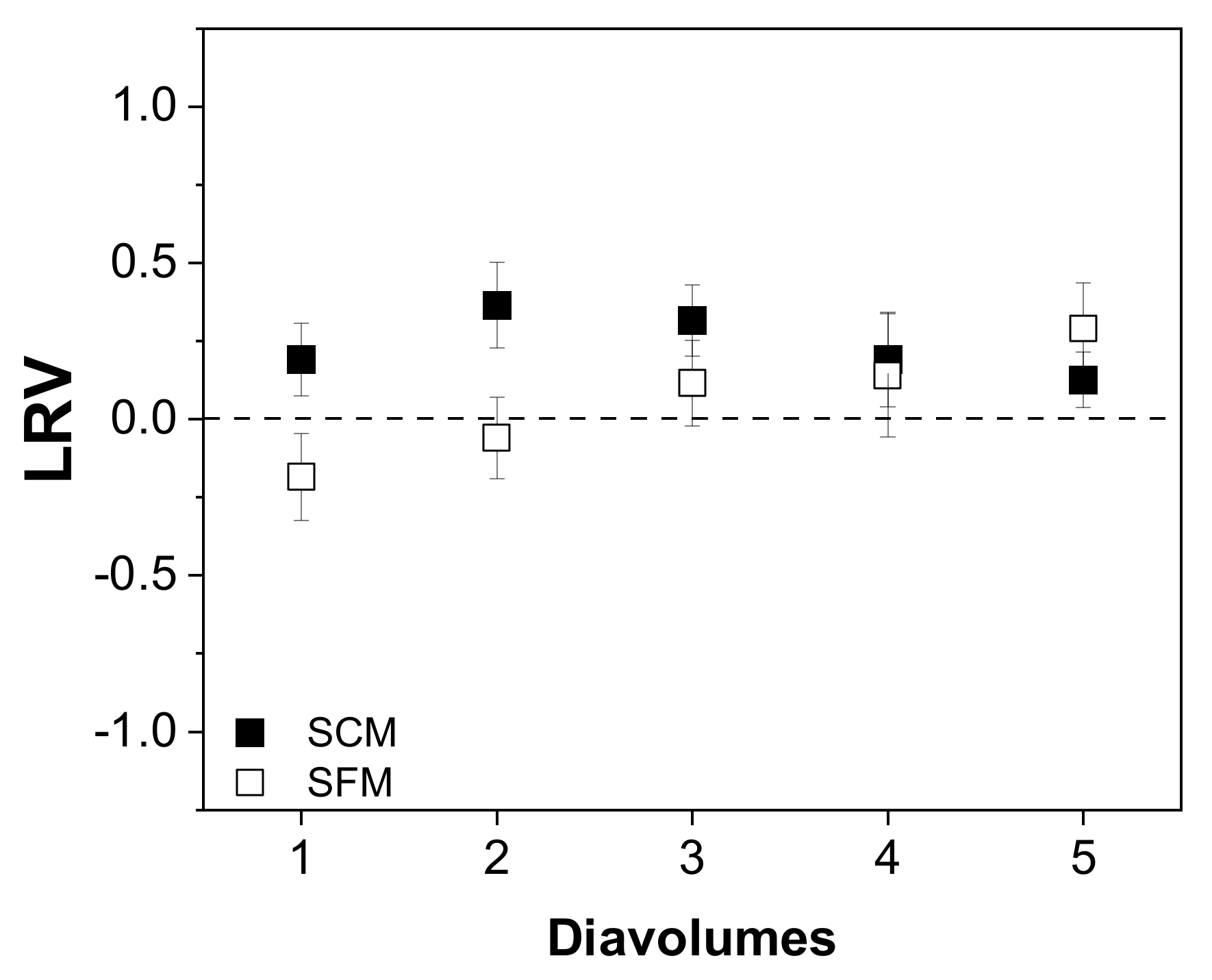
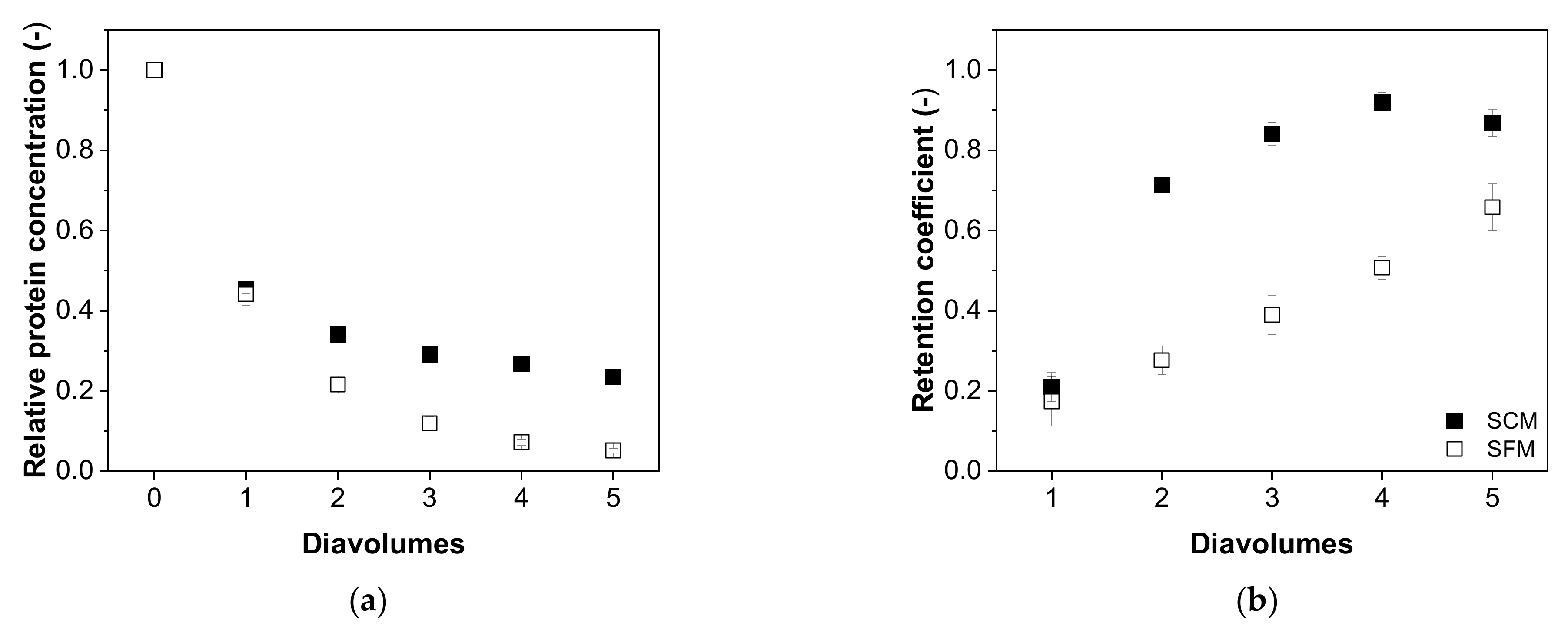
3.4.2. Impurity Depletion
3.5. Summary of Purification Data for MV in SCM and SFM
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Engeland, C.E.; Ungerechts, G. Measles Virus as an Oncolytic Immunotherapy. Cancers 2021, 13, 544. [Google Scholar] [CrossRef] [PubMed]
- Aref, S.; Bailey, K.; Fielding, A. Measles to the Rescue: A Review of Oncolytic Measles Virus. Viruses 2016, 8, 294. [Google Scholar] [CrossRef] [Green Version]
- Msaouel, P.; Opyrchal, M.; Dispenzieri, A.; Peng, K.W.; Federspiel, M.J.; Russell, S.J.; Galanis, E. Clinical Trials with Oncolytic Measles Virus: Current Status and Future Prospects. Curr. Cancer Drug Targets 2018, 18, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, Y.R.; Schulze, A.J. Oncolytic Viruses for Malignant Glioma: On the Verge of Success? Viruses 2021, 13, 1294. [Google Scholar] [CrossRef] [PubMed]
- Mühlebach, M.D. Measles virus in cancer therapy. Curr. Opin. Virol. 2020, 41, 85–97. [Google Scholar] [CrossRef]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loewe, D.; Dieken, H.; Grein, T.A.; Weidner, T.; Salzig, D.; Czermak, P. Opportunities to debottleneck the downstream processing of the oncolytic measles virus. Crit. Rev. Biotechnol. 2020, 40, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Gerstenberger, J.; Salzig, D.; Mühlebach, M.D.; Cichutek, K.; Pörtner, R.; Czermak, P. Oncolytic measles viruses produced at different scales under serum-free conditions. Eng. Life Sci. 2015, 15, 425–436. [Google Scholar] [CrossRef]
- Grein, T.A.; Schwebel, F.; Kress, M.; Loewe, D.; Dieken, H.; Salzig, D.; Weidner, T.; Czermak, P. Screening different host cell lines for the dynamic production of measles virus. Biotechnol. Prog. 2017, 33, 989–997. [Google Scholar] [CrossRef]
- Grein, T.A.; Loewe, D.; Dieken, H.; Salzig, D.; Weidner, T.; Czermak, P. High titer oncolytic measles virus production process by integration of dielectric spectroscopy as online monitoring system. Biotechnol. Bioeng. 2018, 115, 1186–1194. [Google Scholar] [CrossRef]
- Grein, T.A.; Loewe, D.; Dieken, H.; Weidner, T.; Salzig, D.; Czermak, P. Aeration and Shear Stress Are Critical Process Parameters for the Production of Oncolytic Measles Virus. Front. Bioeng. Biotechnol. 2019, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, K.; Salzig, D.; Röder, Y.; Gerstenberger, J.; Mühlebach, M.D.; Cichutek, K.; Pörtner, R.; Czermak, P. Influence of Process Conditions on Measles Virus Stability. Am. J. Biochem. Biotechnol. 2013, 9, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Loewe, D.; Häussler, J.; Grein, T.A.; Dieken, H.; Weidner, T.; Salzig, D.; Czermak, P. Forced Degradation Studies to Identify Critical Process Parameters for the Purification of Infectious Measles Virus. Viruses 2019, 11, 725. [Google Scholar] [CrossRef] [Green Version]
- Sviben, D.; Forčić, D.; Kurtović, T.; Halassy, B.; Brgles, M. Stability, biophysical properties and effect of ultracentrifugation and diafiltration on measles virus and mumps virus. Arch. Virol. 2016, 161, 1455–1467. [Google Scholar] [CrossRef]
- Weiss, K.; Salzig, D.; Mühlebach, M.D.; Cichutek, K.; Pörtner, R.; Czermak, P. Key parameters of Measles virus production for oncoltyic virotherapy. Am. J. Biochem. Biotechnol. 2012, 8, 81–98. [Google Scholar]
- U.S. Food and Drug Administration. Guidance for Industry—Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). FDA. 2020. Available online: https://www.fda.gov/media/113760/download (accessed on 5 January 2022).
- Ungerechts, G.; Bossow, S.; Leuchs, B.; Holm, P.S.; Rommelaere, J.; Coffey, M.; Coffin, R.; Bell, J.; Nettelbeck, D.M. Moving oncolytic viruses into the clinic: Clinical-grade production, purification, and characterization of diverse oncolytic viruses. Mol. Ther. Methods Clin. Dev. 2016, 3, 16018. [Google Scholar] [CrossRef] [Green Version]
- Sviben, D.; Forcic, D.; Ivancic-Jelecki, J.; Halassy, B.; Brgles, M. Recovery of infective virus particles in ion-exchange and hydrophobic interaction monolith chromatography is influenced by particle charge and total-to-infective particle ratio. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1054, 10–19. [Google Scholar] [CrossRef]
- Eckhardt, D.; Dieken, H.; Loewe, D.; Grein, T.A.; Salzig, D.; Czermak, P. Purification of oncolytic measles virus by cation-exchange chromatography using resin-based stationary phases. Sep. Sci. Technol. 2021, 1–11. [Google Scholar] [CrossRef]
- Hoffmann, D.; Leber, J.; Loewe, D.; Lothert, K.; Oppermann, T.; Zitzmann, J.; Weidner, T.; Salzig, D.; Wolff, M.; Czermak, P. Purification of New Biologicals Using Membrane-Based Processes. In Current Trends and Future Developments on (bio-) Membranes: Membrane Processes in the Pharmaceutical and Biotechnological Field; Basile, A.B., Charcosset, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 123–150. ISBN 9780128136065. [Google Scholar]
- Wickramasinghe, S.R.; Kalbfuss, B.; Zimmermann, A.; Thom, V.; Reichl, U. Tangential flow microfiltration and ultrafiltration for human influenza A virus concentration and purification. Biotechnol. Bioeng. 2005, 92, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Michalsky, R.; Passarelli, A.L.; Pfromm, P.H.; Czermak, P. Concentration of the baculovirus Autographa californica M nucleopolyhedrovirus (AcMNPV) by ultrafiltration. Desalination 2010, 250, 1125–1127. [Google Scholar] [CrossRef] [Green Version]
- Grzenia, D.L.; Carlson, J.O.; Wickramasinghe, S.R. Tangential flow filtration for virus purification. J. Membr. Sci. 2008, 321, 373–380. [Google Scholar] [CrossRef]
- Bellini, W.J.; Trudgett, A.; McFarlin, D.E. Purification of Measles Virus with Preservation of Infectivity and Antigenicity. J. Gen. Virol. 1979, 43, 633–639. [Google Scholar] [CrossRef]
- Udem, S.A. Measles virus: Conditions for the propagation and purification of infectious virus in high yield. J. Virol. Methods 1984, 8, 123–136. [Google Scholar] [CrossRef]
- Langfield, K.K.; Walker, H.J.; Gregory, L.C.; Federspiel, M.J. Manufacture of Measles Viruses. In Viral Vectors for Gene Therapy: Methods and Protocols; Merten, O.-W., Al-Rubeai, M., Eds.; Humana Pr: New York, NY, USA, 2011; pp. 345–366. ISBN 978-1-61779-094-2. [Google Scholar]
- Vicente, T.; Peixoto, C.; Carrondo, M.J.T.; Alves, P.M. Purification of recombinant baculoviruses for gene therapy using membrane processes. Gene Ther. 2009, 16, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, C.; Sousa, M.F.Q.; Silva, A.C.; Carrondo, M.J.T.; Alves, P.M. Downstream processing of triple layered rotavirus like particles. J. Biotechnol. 2007, 127, 452–461. [Google Scholar] [CrossRef]
- Carvalho, S.B.; Silva, R.J.S.; Moleirinho, M.G.; Cunha, B.; Moreira, A.S.; Xenopoulos, A.; Alves, P.M.; Carrondo, M.J.T.; Peixoto, C. Membrane-Based Approach for the Downstream Processing of Influenza Virus-Like Particles. Biotechnol. J. 2019, 14, e1800570. [Google Scholar] [CrossRef]
- Nestola, P.; Martins, D.L.; Peixoto, C.; Roederstein, S.; Schleuss, T.; Alves, P.M.; Mota, J.P.B.; Carrondo, M.J.T. Evaluation of novel large cut-off ultrafiltration membranes for adenovirus serotype 5 (Ad5) concentration. PLoS ONE 2014, 9, e115802. [Google Scholar] [CrossRef] [Green Version]
- Kovács, Z.; Czermak, P. Diafiltration. In Encyclopedia of Membrane Science and Technology; Vrijen Hoek, E.M., Tarabara, V.V., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; p. 135. ISBN 9781118522318. [Google Scholar]
- Paulen, R.; Foley, G.; Fikar, M.; Kovács, Z.; Czermak, P. Minimizing the process time for ultrafiltration/diafiltration under gel polarization conditions. J. Membr. Sci. 2011, 380, 148–154. [Google Scholar] [CrossRef]
- Paulen, R.; Fikar, M.; Kovács, Z.; Czermak, P. Process optimization of diafiltration with time-dependent water adding for albumin production. Chem. Eng. Process. Process Intensif. 2011, 50, 815–821. [Google Scholar] [CrossRef]
- Paulen, R.; Fikar, M.; Foley, G.; Kovács, Z.; Czermak, P. Optimal feeding strategy of diafiltration buffer in batch membrane processes. J. Membr. Sci. 2012, 411–412, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, N.; Cheryan, M. Process optimization in ultrafiltration: Flux-time considerations in the purification of macromolecules. Chem. Eng. Commun. 1991, 106, 57–69. [Google Scholar] [CrossRef]
- Loewe, D.; Grein, T.A.; Dieken, H.; Weidner, T.; Salzig, D.; Czermak, P. Tangential Flow Filtration for the Concentration of Oncolytic Measles Virus: The Influence of Filter Properties and the Cell Culture Medium. Membranes 2019, 9, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loewe, D.M. Untersuchung eines Filtrationsbasierten Aufreinigungsprozesses für die APPLIKATION Onkolytischer Masernviren in der Krebstherapie: Charakterisierung des Masernvirus Sowie der Auswirkung des Zellkulturmediums auf den Aufreinigungsprozess; Shaker Verlag: Düren, Germany, 2021; ISBN 978-3-8440-7972-2. [Google Scholar]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Ramakrishnan, M.A. Determination of 50% endpoint titer using a simple formula. World J. Virol. 2016, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- The European Agency for the Evaluation of Medicinal Products. Note for Guidance on Virus Validation Studies: The Design, Contribution and Interpretation of Studies Validating the Inactivation and Removal of Viruses. EMA. 1996. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/note-guidance-virus-validation-studies-design-contribution-interpretation-studies-validating_en.pdf (accessed on 5 January 2022).
- Galanis, E.; Atherton, P.J.; Maurer, M.J.; Knutson, K.L.; Dowdy, S.C.; Cliby, W.A.; Haluska, P.; Long, H.J.; Oberg, A.; Aderca, I.; et al. Oncolytic Measles Virus Expressing the Sodium Iodide Symporter to Treat Drug-Resistant Ovarian Cancer. Cancer Res. 2014, 75, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.J.; Federspiel, M.J.; Peng, K.-W.; Tong, C.; Dingli, D.; Morice, W.G.; Lowe, V.; O’Connor, M.K.; Kyle, R.A.; Leung, N.; et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin. Proc. 2014, 89, 926–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daikoku, E.; Morita, C.; Kohno, T.; Sano, K. Analysis of Morphology and Infectivity of Measles Virus Particles. Bull. Osaka Med. Coll. 2007, 53, 107–114. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measles Virus in | Virus Titer (TCID50 mL−1) | Total RNA (Copies mL−1) | Total RNA/Infectious Particle Titer (RT/I) | Proteins (µg mL−1) | DNA (ng mL−1) |
|---|---|---|---|---|---|
| SCM | 1.6 × 105 ± 3.9 × 104 | 4.9 × 109 ± 8.9 × 108 | 3.1 × 104 | 5071.8 ± 163.1 | 198.0 ± 7.1 |
| SFM | 3.4 × 104 ± 6.9 × 103 | 7.2 × 109 ± 1.8 × 109 | 2.1 × 105 | 528.2 ± 17.3 | 122.6 ± 3.1 |
| Serum-Containing Medium | Serum-Free Medium | ||||
|---|---|---|---|---|---|
| Preconcentration Factor | Mode | Pseudo-Steady-State Flux | Filtration Time | Pseudo-Steady-State Flux | Filtration Time |
| (−) | (L m−2 h−1) | (h) | (L m−2 h−1) | (h) | |
| 1 | Continuous | 25.2 | 3.86 | 51.1 | 1.94 |
| 1.33 | Continuous | 27.1 | 3.03 | 56.9 | 1.44 |
| 2 | Continuous | 24.8 | 2.23 | 56.2 | 1.05 |
| Discontinuous | 26.1 | 2.07 | 65.8 | 0.92 | |
| 4 | Continuous | 26.8 | 1.32 | 62.7 | 0.63 |
| Serum-Containing Medium | Serum-Free Medium | |||||
|---|---|---|---|---|---|---|
| Retention Factor | hcDNA | Total Proteins | Total RNA | hcDNA | Total Proteins | Total RNA |
| Pre–CF: 4 (n = 8) | 0.41 ± 0.10 | 0.29 ± 0.08 | 0.36 ± 0.13 | 0.24 ± 0.08 | 0.11 ± 0.05 | 0.62 ± 0.16 |
| Post–CF: 4 (n = 7) | 0.55 ± 0.08 | 0.52 ± 0.04 | 0.57 ± 0.18 | 0.65 ± 0.13 | 0.20 ± 0.14 | 0.50 ± 0.13 |
| Post–CF: 8 (n = 12) | 0.61 ± 0.05 | 0.48 ± 0.03 | 0.57 ± 0.12 | 0.58 ± 0.09 | 0.15 ± 0.08 | 0.49 ± 0.10 |
| Final Content | ||||
|---|---|---|---|---|
| MV in Serum-Containing Medium | MV in Serum-Free Medium | |||
| Infectious virus | 3.2 × 107 ± 6.2 × 106 TCID50 | 49.3 ± 9.6% | 7.4 × 106 ± 2.7 × 106 TCID50 | 54.5 ± 20.2% |
| Total viral RNA | 1.4 × 1010 ± 1.6 × 109 copies mL−1 | 35.1 ± 4.1% | 1.2 × 1010 ± 2.3 × 109 copies mL−1 | 19.9 ± 4.1% |
| hcDNA | 369.2 ± 19.6 ng mL−1 | 23.3 ± 1.2% | 19.1 ± 5.6 ng mL−1 | 2.0 ± 0.6% |
| Total protein | 2882.1 ± 166.0 µg mL−1 | 7.1 ± 0.4% | 29.4 ± 3.6 µg mL−1 | 0.7 ± 0.1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loewe, D.; Dieken, H.; Grein, T.A.; Salzig, D.; Czermak, P. A Combined Ultrafiltration/Diafiltration Process for the Purification of Oncolytic Measles Virus. Membranes 2022, 12, 105. https://doi.org/10.3390/membranes12020105
Loewe D, Dieken H, Grein TA, Salzig D, Czermak P. A Combined Ultrafiltration/Diafiltration Process for the Purification of Oncolytic Measles Virus. Membranes. 2022; 12(2):105. https://doi.org/10.3390/membranes12020105
Chicago/Turabian StyleLoewe, Daniel, Hauke Dieken, Tanja A. Grein, Denise Salzig, and Peter Czermak. 2022. "A Combined Ultrafiltration/Diafiltration Process for the Purification of Oncolytic Measles Virus" Membranes 12, no. 2: 105. https://doi.org/10.3390/membranes12020105
APA StyleLoewe, D., Dieken, H., Grein, T. A., Salzig, D., & Czermak, P. (2022). A Combined Ultrafiltration/Diafiltration Process for the Purification of Oncolytic Measles Virus. Membranes, 12(2), 105. https://doi.org/10.3390/membranes12020105