
Helicobacter pylori Infection and Chronic Immune Thrombocytopenia


{kind=link}
Abstract
:1. Introduction
2. Immune Thrombocytopenia and H. pylori Infection
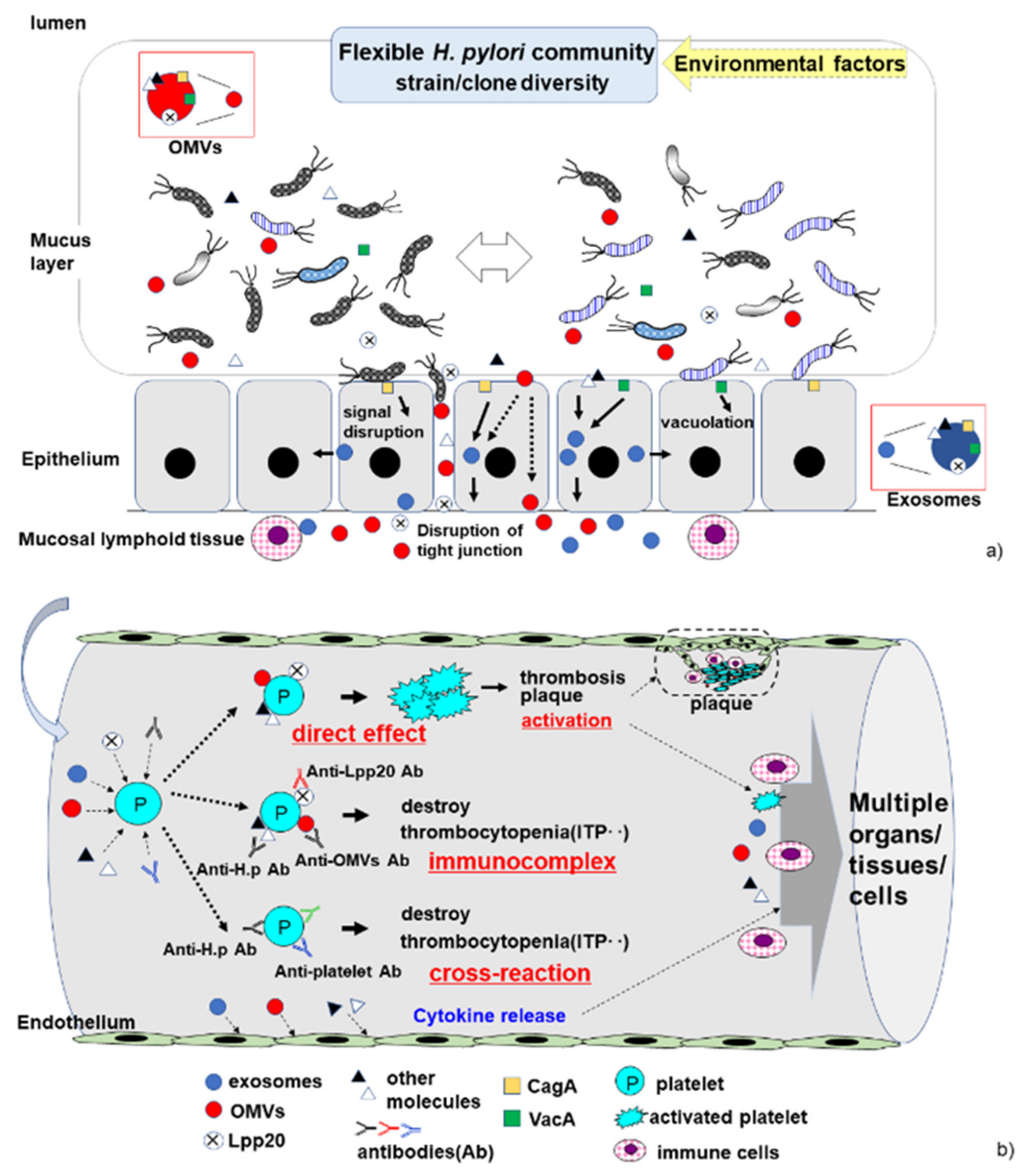
3. Mechanistic Pathway of ITP Development
3.1. Molecular Mimicry and Cross-Reaction
3.2. Platelet Activation and Aggregation
3.3. Host Immune Response to H. pylori Infection
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fischbach, W.; Malfertheiner, P. Helicobacter pylori Infection. Dtsch. Arztebl. Int. 2018, 115, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Zhang, W.; Huang, Y.; Wang, Y.; Shao, Q.; Wu, X.; Lu, N.; Xie, C. Effect of Helicobacter pylori eradication on hyperplastic gastric polyps: A systematic review and meta-analysis. Helicobacter 2021, 26, e12838. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Lee, H.; Kang, M.; Kim, J.E.; Choi, Y.-H.; Min, Y.W.; Min, B.-H.; Lee, J.H.; Son, H.J.; Rhee, P.-L.; et al. Helicobacter pylori is associated with dyslipidemia but not with other risk factors of cardiovascular disease. Sci. Rep. 2016, 6, 38015. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Fujita, A.; Yamazaki, S.; Asaka, M.; Azuma, T.; Hatakeyama, M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc. Natl. Acad. Sci. USA 2002, 99, 14428–14433. [Google Scholar] [CrossRef]
- Ailloud, F.; Didelot, X.; Woltemate, S.; Pfaffinger, G.; Overmann, J.; Bader, R.C.; Schulz, C.; Malfertheiner, P.; Suerbaum, S. Within-host evolution of Helicobacter pylori shaped by niche-specific adaptation, intragastric migrations and selective sweeps. Nat. Commun. 2019, 10, 2273. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kira, M.; Konishi, S.; Uchiyama, J.; Matsuzaki, S.; Matsumura, Y. Polymorphisms in the Helicobacter pylori NY43 strain and its prophage-cured derivatives. Microbiology 2018, 164, 877–882. [Google Scholar] [CrossRef]
- De Korwin, J.-D.; Ianiro, G.; Gibiino, G.; Gasbarrini, A. Helicobacter pylori infection and extragastric diseases in 2017. Helicobacter 2017, 22 (Suppl. S1), e12411. [Google Scholar] [CrossRef]
- Pellicano, R.; Ianiro, G.; Fagoonee, S.; Settanni, C.R.; Gasbarrini, A. Review: Extragastric diseases and Helicobacter pylori. Helicobacter 2020, 25 (Suppl. S1), e12741. [Google Scholar] [CrossRef]
- Qiang, L.; Hu, J.; Tian, M.; Li, Y.; Ren, C.; Deng, Y.; Jiang, Y. Extracellular vesicles from Helicobacter pylori-infected cells and Helicobacter pylori outer membrane vesicles in atherosclerosis. Helicobacter 2022, 27, e12877. [Google Scholar] [CrossRef]
- Franceschi, F.; Zuccalà, G.; Roccarina, D.; Gasbarrini, A. Clinical effects of Helicobacter pylori outside the stomach. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 234–242. [Google Scholar] [CrossRef]
- Lee, M.; Baek, H.; Park, J.S.; Kim, S.; Kyung, C.; Baik, S.J.; Lee, B.K.; Kim, J.-H.; Ahn, C.W.; Kim, K.R.; et al. Current Helicobacter pylori infection is significantly associated with subclinical coronary atherosclerosis in healthy subjects: A cross-sectional study. PLoS ONE 2018, 13, e0193646. [Google Scholar] [CrossRef]
- Yokota, K.; Osaki, T.; Hayashi, S.; Yokota, S.; Takeuchi, H.; Rimbara, E.; Ojima, H.; Sato, T.; Yonezawa, H.; Shibayama, K.; et al. Establishment of a reference panel of Helicobacter pylori strains for antimicrobial susceptibility testing. Helicobacter 2022, 27, e12874. [Google Scholar] [CrossRef]
- Neunert, C.E. Current management of immune thrombocytopenia. Hematology 2013, 2013, 276–282. [Google Scholar] [CrossRef]
- Semple, J.W.; Rebetz, J.; Maouia, A.; Kapur, R. An update on the pathophysiology of immune thrombocytopenia. Curr. Opin. Hematol. 2020, 27, 423–429. [Google Scholar] [CrossRef]
- Gasbarrini, A.; Franceschi, F.; Tartaglione, R.; Landolfi, R.; Pola, P.; Gasbarrini, G. Regression of autoimmune thrombocytopenia after eradication of Helicobacter pylori. Lancet 1998, 352, 878. [Google Scholar] [CrossRef]
- O’Neill, C.M.; Weitz, I.C.; O’Connell, C.; Liebman, H.A. Ethnic and racial difference in Helicobacter pylori infection in patients with immune thrombocytopenia treated at a major urban medical center. Platelets 2019, 30, 413–417. [Google Scholar] [CrossRef]
- Frydman, G.H.; Davis, N.; Beck, P.L.; Fox, J.G. Helicobacter pylori Eradication in Patients with Immune Thrombocytopenic Purpura: A Review and the Role of Biogeography. Helicobacter 2015, 20, 239–251. [Google Scholar] [CrossRef]
- Vishnu, P.; Duncan, J.; Connell, N.; Cooper, N.; Lim, W.; Rodeghiero, F.; Tomiyama, Y.; Grace, R.F.; Bakchoul, T.; Arnold, D.M.; et al. International survey on Helicobacter pylori testing in patients with immune thrombocytopenia: Communication of the platelet immunology scientific and standardization committee. J. Thromb. Haemost. 2021, 19, 287–296. [Google Scholar] [CrossRef]
- Pezeshki, S.M.S.; Saki, N.; Ghandali, M.V.; Ekrami, A.; Avarvand, A.Y. Effect of Helicobacter pylori eradication on patients with ITP: A meta-analysis of studies conducted in the Middle East. Blood Res. 2021, 56, 38–43. [Google Scholar] [CrossRef]
- Amedei, A.; Bergman, M.P.; Appelmelk, B.J.; Azzurri, A.; Benagiano, M.; Tamburini, C.; van der Zee, R.; Telford, J.L.; Vandenbroucke-Grauls, C.M.J.E.; D’Elios, M.M.; et al. Molecular Mimicry between Helicobacter pylori Antigens and H+,K+–Adenosine Triphosphatase in Human Gastric Autoimmunity. J. Exp. Med. 2003, 198, 1147–1156. [Google Scholar] [CrossRef]
- Faller, G.; Winter, M.; Steininger, H.; Lehn, N.; Meining, A.; Bayerdörffer, E.; Kirchner, T. Decrease of Antigastric Autoantibodies in Helicobacter pylori Gastritis after Cure of Infection. Pathol. Res. Pract. 1999, 195, 243–246. [Google Scholar] [CrossRef]
- Wang, L.; Cao, Z.-M.; Zhang, L.-L.; Dai, X.-C.; Liu, Z.-J.; Zeng, Y.-X.; Li, X.-Y.; Wu, Q.-J.; Lv, W.-L. Helicobacter pylori and Autoimmune Diseases: Involving Multiple Systems. Front. Immunol. 2022, 13, 833424. [Google Scholar] [CrossRef] [PubMed]
- Ihtesham, A.; Maqbool, S.; Nadeem, M.; Janjua, M.B.A.; Sundus, O.; Naqqash, A.B.; Mohamed, W.I.; Haider, S.T.; Ahmad, M.; Mustafa, M.A.T.; et al. Helicobacter pylori induced Immune Thrombocytopenic Purpura and perspective role of Helicobacter pylori eradication therapy for treating Immune Thrombocytopenic Purpura. AIMS Microbiol. 2021, 7, 284–303. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Khellaf, M.; Desforges, L.; Lee, K.; Schaeffer, A.; Godeau, B.; Bierling, P. Autoimmune Thrombocytopenic Purpura and Helicobacter pylori Infection. Arch. Intern. Med. 2002, 162, 1033–1036. [Google Scholar] [CrossRef]
- Kim, B.J.; Kim, H.S.; Jang, H.J.; Kim, J.H. Helicobacter pylori Eradication in Idiopathic Thrombocytopenic Purpura: A Meta-Analysis of Randomized Trials. Gastroenterol. Res. Pract. 2018, 2018, 6090878. [Google Scholar] [CrossRef]
- Takahashi, T.; Yujiri, T.; Shinohara, K.; Inoue, Y.; Sato, Y.; Fujii, Y.; Okubo, M.; Zaitsu, Y.; Ariyoshi, K.; Nakamura, Y.; et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori -associated chronic idiopathic thrombocytopenic purpura. Br. J. Haematol. 2004, 124, 91–96. [Google Scholar] [CrossRef]
- Kodama, M.; Kitadai, Y.; Ito, M.; Kai, H.; Masuda, H.; Tanaka, S.; Yoshihara, M.; Fujimura, K.; Chayama, K. Immune Response to CagA Protein is Associated with Improved Platelet Count after Helicobacter pylori Eradication in Patients with Idiopathic Thrombocytopenic Purpura. Helicobacter 2007, 12, 36–42. [Google Scholar] [CrossRef]
- Satoh, K.; Hirayama, T.; Takano, K.; Suzuki-Inoue, K.; Sato, T.; Ohta, M.; Nakagomi, J.; Ozaki, Y. VacA, the vacuolating cytotoxin of Helicobacter pylori, binds to multimerin 1 on human platelets. Thromb. J. 2013, 11, 23. [Google Scholar] [CrossRef]
- Franceschi, F.; Christodoulides, N.; Kroll, M.H.; Genta, R.M. Helicobacter pylori and Idiopathic Thrombocytopenic Purpura. Ann. Intern. Med. 2004, 140, 766–767. [Google Scholar] [CrossRef]
- Shimoda, A.; Ueda, K.; Nishiumi, S.; Murata-Kamiya, N.; Mukai, S.-A.; Sawada, S.-I.; Azuma, T.; Hatakeyama, M.; Akiyoshi, K. Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci. Rep. 2016, 6, 18346. [Google Scholar] [CrossRef]
- Jackson, S.; Beck, P.L.; Pineo, G.F.; Poon, M.-C. Helicobacter pylori eradication: Novel therapy for immune thrombocytopenic purpura? A review of the literature. Am. J. Hematol. 2005, 78, 142–150. [Google Scholar] [CrossRef]
- Morimoto, N.; Takeuchi, H.; Takahashi, T.; Ueta, T.; Tanizawa, Y.; Kumon, Y.; Kobayashi, M.; Sugiura, T. Helicobacter pylori-associated chronic idiopathic thrombocytopenic purpura and low molecular weight H. pylori proteins. Scand. J. Infect. Dis. 2007, 39, 409–416. [Google Scholar] [CrossRef]
- Takeuchi, H.; Islam, J.; Kaneko, A.; Kimura, A.; Shida, T.; Oboshi, W.; Katayama, H.; Oishi, T.; Fujieda, M.; Morimoto, N. Helicobacter pylori protein that binds to and activates platelet specifically reacts with sera of H. pylori-associated chronic immune thrombocytopenia. Platelets 2021, 32, 1120–1123. [Google Scholar] [CrossRef]
- Cao, P.; McClain, M.S.; Forsyth, M.H.; Cover, T.L. Extracellular Release of Antigenic Proteins by Helicobacter pylori. Infect. Immun. 1998, 66, 2984–2986. [Google Scholar] [CrossRef]
- Vallese, F.; Mishra, N.M.; Pagliari, M.; Berto, P.; Codolo, G.; de Bernard, M.; Zanotti, G. Helicobacter pylori antigenic Lpp20 is a structural homologue of Tipα and promotes epithelial-mesenchymal transition. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 3263–3271. [Google Scholar] [CrossRef]
- Kostrzynska, M.; O’Toole, P.W.; Taylor, D.E.; Trust, T.J. Molecular characterization of a conserved 20-kilodalton membrane-associated lipoprotein antigen of Helicobacter pylori. J. Bacteriol. 1994, 176, 5938–5948. [Google Scholar] [CrossRef]
- Byrne, M.F.; Kerrigan, S.W.; Corcoran, P.A.; Atherton, J.C.; Murray, F.E.; Fitzgerald, D.J.; Cox, D.M. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology 2003, 124, 1846–1854. [Google Scholar] [CrossRef]
- Riad, M. Association of Helicobacter pylori infection with coronary artery disease: Is it an independent risk factor? Egypt. Heart J. 2021, 73, 61. [Google Scholar] [CrossRef]
- Scopel-Guerra, A.; Olivera-Severo, D.; Staniscuaski, F.; Uberti, A.F.; Callai-Silva, N.; Jaeger, N.; Porto, B.N.; Carlini, C.R. The Impact of Helicobacter pylori Urease upon Platelets and Consequent Contributions to Inflammation. Front. Microbiol. 2017, 8, 2447. [Google Scholar] [CrossRef]
- Kowalski, M.; Rees, W.; Konturek, P.; Grove, R.; Scheffold, T.; Meixner, H.; Brunec, M.; Franz, N.; Konturek, J.; Pieniazek, P.; et al. Detection of Helicobacter pylori specific DNA in human atheromatous coronary arteries and its association to prior myocardial infarction and unstable angina. Dig. Liver Dis. 2002, 34, 398–402. [Google Scholar] [CrossRef]
- Li, N.; Liu, S.-F.; Dong, K.; Zhang, G.-C.; Huang, J.; Wang, Z.-H.; Wang, T.-J. Exosome-Transmitted miR-25 Induced by H. pylori Promotes Vascular Endothelial Cell Injury by Targeting KLF2. Front. Cell. Infect. Microbiol. 2019, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, P.A.; Atherton, J.C.; Kerrigan, S.W.; Wadstrom, T.; Murray, F.E.; Peek, R.M.; Fitzgerald, D.J.; Cox, D.M.; Byrne, M.F. The effect of different strains of Helicobacter pylori on platelet aggregation. Can. J. Gastroenterol. 2007, 21, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Asahi, A.; Nishimoto, T.; Okazaki, Y.; Suzuki, H.; Masaoka, T.; Kawakami, Y.; Ikeda, Y.; Kuwana, M. Helicobacter pylori eradication shifts monocyte Fcγ receptor balance toward inhibitory FcγRIIB in immune thrombocytopenic purpura patients. J. Clin. Investig. 2008, 118, 2939–2949. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, S.; Iizumi, T.; Watanabe, E.; Shimizu, M.; Kamiya, S.; Nagata, K.; Kumagai, Y.; Fukunaga, Y.; Takahashi, H. Implications for Induction of Autoimmunity via Activation of B-1 Cells by Helicobacter pylori Urease. Infect. Immun. 2006, 74, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Yokohama, A.; Osaki, Y.; Ogawa, Y.; Nakahashi, H.; Toyama, K.; Mitsui, T.; Hashimoto, Y.; Koiso, H.; Uchiumi, H.; et al. Circulating plasmacytoid dendritic cells in patients with primary and Helicobacter pylori-associated immune thrombocytopenia. Eur. J. Haematol. 2012, 88, 340–349. [Google Scholar] [CrossRef]
- Appelmelk, B.J.; van Die, I.; van Vliet, S.J.; Vandenbroucke-Grauls, C.M.J.E.; Geijtenbeek, T.B.H.; van Kooyk, Y. Cutting Edge: Carbohydrate Profiling Identifies New Pathogens That Interact with Dendritic Cell-Specific ICAM-3-Grabbing Nonintegrin on Dendritic Cells. J. Immunol. 2003, 170, 1635–1639. [Google Scholar] [CrossRef]
- Voland, P.; Hafsi, N.; Zeitner, M.; Laforsch, S.; Wagner, H.; Prinz, C. Antigenic Properties of HpaA and Omp18, Two Outer Membrane Proteins of Helicobacter pylori. Infect. Immun. 2003, 71, 3837–3843. [Google Scholar] [CrossRef]
- Tummuru, M.K.; Sharma, S.A.; Blaser, M.J. Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Mol. Microbiol. 1995, 18, 867–876. [Google Scholar] [CrossRef]
- Asahi, M.; Azuma, T.; Ito, S.; Ito, Y.; Suto, H.; Nagai, Y.; Tsubokawa, M.; Tohyama, Y.; Maeda, S.; Omata, M.; et al. Helicobacter pylori Caga Protein Can Be Tyrosine Phosphorylated in Gastric Epithelial Cells. J. Exp. Med. 2000, 191, 593–602. [Google Scholar] [CrossRef]
- Holland, R.L.; Bosi, K.D.; Harpring, G.H.; Luo, J.; Wallig, M.; Phillips, H.; Blanke, S.R. Chronic in vivo exposure to Helicobacter pylori VacA: Assessing the efficacy of automated and long-term intragastric toxin infusion. Sci. Rep. 2020, 10, 9307. [Google Scholar] [CrossRef]
- Neunert, C.; Terrell, D.R.; Arnold, D.M.; Buchanan, G.; Cines, D.B.; Cooper, N.; Cuker, A.; Despotovic, J.M.; George, J.N.; Grace, R.F.; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3, 3829–3866. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner, P.; Megraud, F.; O’Morain, C.A.; Gisbert, J.P.; Kuipers, E.J.; Axon, A.T.; Bazzoli, F.; Gasbarrini, A.; Atherton, J.; Graham, D.Y.; et al. Management of Helicobacter pylori infection—The Maastricht V/Florence Consensus Report. Gut 2017, 66, 6–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Jung, H.-K.; Lee, H.L.; Jang, J.Y.; Lee, H.; Kim, C.G.; Shin, W.G.; Shin, E.S.; Lee, Y.C. Guidelines for the diagnosis and treatment of Helicobacter pylori infection in Korea, 2013 revised edition. Korean J. Gastroenterol. 2013, 62, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, H.; Kuwana, M.; Hato, T.; Takafuta, T.; Fujimura, K.; Kurata, Y.; Murata, M.; Tomiyama, Y. Reference guide for management of adult immune thrombocytopenia in Japan: 2019 Revision. Int. J. Hematol. 2020, 111, 329–351. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeuchi, H.; Okamoto, A. Helicobacter pylori Infection and Chronic Immune Thrombocytopenia. J. Clin. Med. 2022, 11, 4822. https://doi.org/10.3390/jcm11164822
Takeuchi H, Okamoto A. Helicobacter pylori Infection and Chronic Immune Thrombocytopenia. Journal of Clinical Medicine. 2022; 11(16):4822. https://doi.org/10.3390/jcm11164822
Chicago/Turabian StyleTakeuchi, Hiroaki, and Aoi Okamoto. 2022. "Helicobacter pylori Infection and Chronic Immune Thrombocytopenia" Journal of Clinical Medicine 11, no. 16: 4822. https://doi.org/10.3390/jcm11164822
APA StyleTakeuchi, H., & Okamoto, A. (2022). Helicobacter pylori Infection and Chronic Immune Thrombocytopenia. Journal of Clinical Medicine, 11(16), 4822. https://doi.org/10.3390/jcm11164822