
New Trends and Therapies for Familial Hypercholesterolemia


Abstract
:1. New Trends in Genetics, Epidemiology, and Atherosclerotic Cardiovascular Disease Risk in Familial Hypercholesterolemia
1.1. Genetics of FH
1.2. Epidemiology
1.3. Heterogeneity in Atherosclerotic Cardiovascular Disease Risk
1.4. Impact of Statin Therapy and Ezetimibe on LDL-C and ASCVD Risk in FH
2. New Trends in Therapies for FH
2.1. PCSK9 Inhibitors
2.2. Bempedoic Acid
2.3. Angiopoietin-like 3 Protein (ANGPTL3) Inhibitors
2.4. MTP Inhibitors
2.5. Gene Therapies
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khera, A.V.; Won, H.-H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients with Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 2578–2589. [Google Scholar] [CrossRef] [PubMed]
- Trinder, M.; Paquette, M.; Cermakova, L.; Ban, M.R.; Hegele, R.A.; Baass, A.; Brunham, L.R. Polygenic Contribution to Low-Density Lipoprotein Cholesterol Levels and Cardiovascular Risk in Monogenic Familial Hypercholesterolemia. Circ. Genom. Precis. Med. 2020, 13, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.D.; Gidding, S.S.; Hegele, R.A.; Cuchel, M.A.; Barter, P.J.; Watts, G.F.; Baum, S.J.; Catapano, A.L.; Chapman, M.J.; Defesche, J.C.; et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: A consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016, 4, 850–861. [Google Scholar] [CrossRef]
- Gallo, A.; de Isla, L.P.; Charrière, S.; Vimont, A.; Alonso, R.; Muñiz-Grijalvo, O.; Díaz-Díaz, J.L.; Zambón, D.; Moulin, P.; Bruckert, E.; et al. The Added Value of Coronary Calcium Score in Predicting Cardiovascular Events in Familial Hypercholesterolemia. JACC Cardiovasc. Imaging 2021, 14, 2414–2424. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Wang, M.; Homburger, J.R.; Patel, A.P.; Bick, A.G.; Neben, C.L.; Lai, C.; Brockman, D.; Philippakis, A.; Ellinor, P.T.; et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat. Commun. 2020, 11, 3635. [Google Scholar] [CrossRef] [PubMed]
- Miname, M.H.; Bittencourt, M.S.; Moraes, S.R.; Alves, R.I.; Silva, P.R.; Jannes, C.E.; Pereira, A.C.; Krieger, J.E.; Nasir, K.; Santos, R.D. Coronary Artery Calcium and Cardiovascular Events in Patients with Familial Hypercholesterolemia Receiving Standard Lipid-Lowering Therapy. JACC Cardiovasc. Imaging 2019, 12, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Perez de Isla, L.; Alonso, R.; Mata, N.; Fernández-Pérez, C.; Muñiz, O.; Díaz-Díaz, J.L.; Saltijeral, A.; Fuentes-Jiménez, F.; De Andrés, R.; Zambón, D.; et al. Predicting Cardiovascular Events in Familial Hypercholesterolemia: The SAFEHEART Registry (Spanish Familial Hypercholesterolemia Cohort Study). Circulation 2017, 135, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; Bernard, S.; Cariou, B.; Hegele, R.A.; Genest, J.; Trinder, M.; Brunham, L.R.; Béliard, S.; Baass, A. Familial Hypercholesterolemia-Risk-Score: A New Score Predicting Cardiovascular Events and Cardiovascular Mortality in Familial Hypercholesterolemia. Arter. Thromb. Vasc. Biol. 2021, 41, 2632–2640. [Google Scholar] [CrossRef]
- Bélanger, A.M.; Akioyamen, L.E.; Ruel, I.; Hales, L.; Genest, J. Aortic stenosis in homozygous familial hypercholesterolaemia: A paradigm shift over a century. Eur. Heart J. 2022, 43, 3227–3239. [Google Scholar] [CrossRef]
- Thompson, G.R.; Blom, D.J.; Marais, A.D.; Seed, M.; Pilcher, G.J.; Raal, F.J. Survival in homozygous familial hypercholesterolaemia is determined by the on-treatment level of serum cholesterol. Eur. Heart J. 2018, 39, 1162–1168. [Google Scholar] [CrossRef]
- Raal, F.J.; Pilcher, G.J.; Panz, V.R.; van Deventer, H.E.; Brice, B.C.; Blom, D.J.; Marais, A.D. Reduction in Mortality in Subjects with Homozygous Familial Hypercholesterolemia Associated with Advances in Lipid-Lowering Therapy. Circulation 2011, 124, 2202–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versmissen, J.; Oosterveer, D.; Yazdanpanah, M.; Defesche, J.C.; Basart, D.C.G.; Liem, A.H.; Heeringa, J.; Witteman, J.C.; Lansberg, P.J.; Kastelein, J.J.P.; et al. Efficacy of statins in familial hypercholesterolaemia: A long term cohort study. BMJ Clin. Res. Ed. 2008, 337, a2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besseling, J.; Hovingh, G.K.; Huijgen, R.; Kastelein, J.J.P.; Hutten, B.A. Statins in Familial Hypercholesterolemia: Consequences for Coronary Artery Disease and All-Cause Mortality. J. Am. Coll. Cardiol. 2016, 68, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Mszar, R.; Nasir, K.; Santos, R.D. Coronary Artery Calcification in Familial Hypercholesterolemia: An Opportunity for Risk Assessment and Shared Decision Making with the Power of Zero? Circulation 2020, 142, 1405–1407. [Google Scholar] [CrossRef]
- Blom, D.J.; Averna, M.; Meagher, E.A.; Theron, H.D.T.; Sirtori, C.R.; Hegele, R.A.; Shah, P.K.; Gaudet, D.; Stefanutti, C.; Vigna, G.; et al. Long-Term Efficacy and Safety of the Microsomal Triglyceride Transfer Protein Inhibitor Lomitapide in Patients with Homozygous Familial Hypercholesterolemia. Circulation 2017, 136, 332–335. [Google Scholar] [CrossRef] [Green Version]
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.-C.; Gipe, D.A.; et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 711–720. [Google Scholar] [CrossRef]
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Primers 2017, 3, 17093. [Google Scholar] [CrossRef]
- Rocha, V.Z.; Santos, R.D. Past, Present, and Future of Familial Hypercholesterolemia Management. Methodist DeBakey Cardiovasc. J. 2021, 17, 28–35. [Google Scholar] [CrossRef]
- Loaiza, N.; Hartgers, M.L.; Reeskamp, L.F.; Balder, J.W.; Rimbert, A.; Bazioti, V.; Wolters, J.C.; Winkelmeijer, M.; Jansen, H.P.; Dallinga-Thie, G.M.; et al. Taking One Step Back in Familial Hypercholesterolemia: STAP1 Does Not Alter Plasma LDL (Low-Density Lipoprotein) Cholesterol in Mice and Humans. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 973–985. [Google Scholar] [CrossRef]
- Tada, M.T.; Rocha, V.Z.; Lima, I.R.; Oliveira, T.G.M.; Chacra, A.P.; Miname, M.H.; Nunes, V.S.; Nakandakare, E.R.; Castelo, M.H.C.G.; Jannes, C.E.; et al. Screening of ABCG5 and ABCG8 Genes for Sitosterolemia in a Familial Hypercholesterolemia Cascade Screening Program. Circ. Genom. Precis. Med. 2022, 15, e003390. [Google Scholar] [CrossRef]
- Talmud, P.J.; Shah, S.; Whittall, R.; Futema, M.; Howard, P.; Cooper, J.A.; Harrison, S.C.; Li, K.; Drenos, F.; Karpe, F.; et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study. Lancet 2013, 381, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Beheshti, S.; Madsen, C.; Varbo, A.; Nordestgaard, B. Worldwide prevalence of familial hypercholesterolemia: Meta-analyses of 11 million subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Vaz, A.J.; Ray, K.K. Epidemiology of familial hypercholesterolaemia: Community and clinical. Atherosclerosis 2018, 277, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhabib, K.F.; Al-Rasadi, K.; Almigbal, T.H.; Batais, M.A.; Al-Zakwani, I.; Al-Allaf, F.A.; Al-Waili, K.; Zadjali, F.; Alghamdi, M.; Alnouri, F.; et al. Familial Hypercholesterolemia in the Arabian Gulf Region: Clinical results of the Gulf FH Registry. PLoS ONE 2021, 16, e0251560. [Google Scholar] [CrossRef] [PubMed]
- Bilen, O.; Pokharel, Y.; Ballantyne, C.M. Genetic Testing in Hyperlipidemia. Cardiol. Clin. 2015, 33, 267–275. [Google Scholar] [CrossRef]
- Villa, G.; Wong, B.; Kutikova, L.; Ray, K.K.; Mata, P.; Bruckert, E. Prediction of cardiovascular risk in patients with familial hypercholesterolaemia. Eur. Heart J. Qual. Care Clin. Outcomes 2017, 3, 274–280. [Google Scholar] [CrossRef]
- Collaboration EASFHS. Global perspective of familial hypercholesterolaemia: A cross-sectional study from the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Lancet 2021, 398, 1713–1725. [Google Scholar] [CrossRef]
- Tromp, T.R.; Hartgers, M.L.; Hovingh, G.K.; Vallejo-Vaz, A.J.; Ray, K.K.; Soran, H.; Freiberger, T.; Bertolini, S.; Harada-Shiba, M.; Blom, D.J.; et al. Worldwide experience of homozygous familial hypercholesterolaemia: Retrospective cohort study. Lancet 2022, 399, 719–728. [Google Scholar] [CrossRef]
- Alves, A.C.; Alonso, R.; Diaz-Diaz, J.L.; Medeiros, A.M.; Jannes, C.E.; Merchan, A.; Vasques-Cardenas, N.A.; Cuevas, A.; Chacra, A.P.; Krieger, J.E.; et al. Phenotypical, Clinical, and Molecular Aspects of Adults and Children with Homozygous Familial Hypercholesterolemia in Iberoamerica. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2508–2515. [Google Scholar]
- Miname, M.H.; Santos, R.D. Reducing cardiovascular risk in patients with familial hypercholesterolemia: Risk prediction and lipid management. Prog. Cardiovasc. Dis. 2019, 62, 414–422. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, e285–e350. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.D.; Stein, E.A.; Hovingh, G.K.; Blom, D.J.; Soran, H.; Watts, G.F.; López, J.A.G.; Bray, S.; Kurtz, C.E.; Hamer, A.W.; et al. Long-Term Evolocumab in Patients with Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2020, 75, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.; Ginsberg, H.N.; Langslet, G.; Hovingh, G.K.; Ceska, R.; Dufour, R.; Blom, D.; Civeira, F.; Krempf, M.; Lorenzato, C.; et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur. Heart J. 2015, 36, 2996–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Santos, R.D.; Ruzza, A.; Hovingh, G.K.; Wiegman, A.; Mach, F.; Kurtz, C.E.; Hamer, A.; Bridges, I.; Bartuli, A.; Bergeron, J.; et al. Cognitive function with evolocumab in pediatric heterozygous familial hypercholesterolemia. N. Engl. J. Med. 2020, 383, 1317–1327. [Google Scholar] [CrossRef]
- Santos, R.D.; Ruzza, A.; Hovingh, G.K.; Stefanutti, C.; Mach, F.; Descamps, O.S.; Bergeron, J.; Wang, B.; Bartuli, A.; Buonuomo, P.S.; et al. Paediatric patients with heterozygous familial hypercholesterolaemia treated with evolocumab for 80 weeks (HAUSER-OLE): A single-arm, multicentre, open-label extension of HAUSER-RCT. Lancet Diabetes Endocrinol. 2022, 10, 732–740. [Google Scholar] [CrossRef]
- Perez de Isla, L.; Alonso, R.; Watts, G.F.; Mata, N.; Saltijeral Cerezo, A.; Muñiz, O.; Fuentes, F.; Diaz-Diaz, J.L.; de Andrés, R.; Zambón, D.; et al. Attainment of LDL Cholesterol Treatment Goals in Patients with Familial Hypercholesterolemia at 5-year Follow-up: SAFEHEART Registry. J. Am. Coll. Cardiol. 2016, 67, 1278–1285. [Google Scholar] [CrossRef]
- Miname, M.H.; Bittencourt, M.S.; Pereira, A.C.; Jannes, C.E.; Krieger, J.E.; Nasir, K.; Santos, R.D. Vascular age derived from coronary artery calcium score on the risk stratification of individuals with heterozygous familial hypercholesterolaemia. Eur. Heart J. Cardiovasc. Imaging 2019, 21, 251–257. [Google Scholar] [CrossRef]
- Mszar, R.; Grandhi, G.R.; Valero-Elizondo, J.; Virani, S.S.; Blankstein, R.; Blaha, M.; Mata, P.; Miname, M.H.; Al Rasadi, K.; Krumholz, H.M.; et al. Absence of Coronary Artery Calcification in Middle-Aged Familial Hypercholesterolemia Patients without Atherosclerotic Cardiovascular Disease. JACC Cardiovasc. Imaging 2020, 13, 1090–1092. [Google Scholar] [CrossRef]
- Santos, R.D.; Shapiro, M.D. Coronary Artery Calcification and Risk Stratification in Familial Hypercholesterolemia: Moving Forward but Not There Yet. JACC Cardiovasc. Imaging 2021, 14, 2425–2428. [Google Scholar] [CrossRef] [PubMed]
- Blom, D.J.; Harada-Shiba, M.; Rubba, P.; Gaudet, D.; Kastelein, J.J.; Charng, M.J.; Pordy, R.; Donahue, S.; Ali, S.; Dong, Y.; et al. Efficacy and Safety of Alirocumab in Adults with Homozygous Familial Hypercholesterolemia: The ODYSSEY HoFH Trial. J. Am. Coll. Cardiol. 2020, 76, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Stefanutti, C.; Julius, U.; Watts, G.F.; Harada-Shiba, M.; Cossu, M.; Schettler, V.J.; De Silvestro, G.; Soran, H.; Van Lennep, J.R.; Pisciotta, L.; et al. Toward an international consensus—Integrating lipoprotein apheresis and new lipid-lowering drugs. J. Clin. Lipidol. 2017, 11, 858–871.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanutti, C.; Thompson, G.R. Lipoprotein Apheresis in the Management of Familial Hypercholesterolaemia: Historical Perspective and Recent Advances. Curr. Atheroscler. Rep. 2015, 17, 465. [Google Scholar] [CrossRef] [PubMed]
- Underberg, J.A.; Cannon, C.P.; Larrey, D.; Makris, L.; Blom, D.; Phillips, H. Long-term safety and efficacy of lomitapide in patients with homozygous familial hypercholesterolemia: Five-year data from the Lomitapide Observational Worldwide Evaluation Registry (LOWER). J. Clin. Lipidol. 2020, 14, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.B. Benefit of early initiation of statins for FH. Nat. Rev. Cardiol. 2020, 17, 8. [Google Scholar] [CrossRef]
- Santos, R.D. Expression of LDLRs (Low-Density Lipoprotein Receptors), Dyslipidemia Severity, and Response to PCSK9 (Proprotein Convertase Subtilisin Kexin Type 9) Inhibition in Homozygous Familial Hypercholesterolemia: Connecting the Dots. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 481–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginsberg, H.N. REVIEW: Efficacy and Mechanisms of Action of Statins in the Treatment of Diabetic Dyslipidemia. J. Clin. Endocrinol. Metab. 2006, 91, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Luirink, I.K.; Wiegman, A.; Kusters, D.M.; Hof, M.H.; Groothoff, J.W.; de Groot, E.; Kastelein, J.J.; Hutten, B.A. 20-Year Follow-up of Statins in Children with Familial Hypercholesterolemia. N. Engl. J. Med. 2019, 381, 1547–1556. [Google Scholar] [CrossRef]
- Garber, A.J.; Abrahamson, M.J.; Barzilay, J.I.; Blonde, L.; Bloomgarden, Z.T.; Bush, M.A.; Dagogo-Jack, S.; DeFronzo, R.A.; Einhorn, D.; Fonseca, V.A.; et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes Management Algorithm—2019 Executive Summary. Endocr. Pract. 2019, 25, 69–101. [Google Scholar] [CrossRef] [Green Version]
- Sugizaki, T.; Watanabe, M.; Horai, Y.; Kaneko-Iwasaki, N.; Arita, E.; Miyazaki, T.; Morimoto, K.; Honda, A.; Irie, J.; Itoh, H. The Niemann-Pick C1 Like 1 (NPC1L1) Inhibitor Ezetimibe Improves Metabolic Disease Via Decreased Liver X Receptor (LXR) Activity in Liver of Obese Male Mice. Endocrinology 2014, 155, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.; Stender, S.; Mata, P.; Sager, P.; Ponsonnet, D.; Melani, L.; Lipka, L.; Suresh, R.; MacCubbin, D.; Veltri, E. Achieving lipoprotein goals in patients at high risk with severe hypercholesterolemia: Efficacy and safety of ezetimibe co-administered with atorvastatin. Am. Heart J. 2004, 148, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Bruckert, E.; Ginsberg, H.N.; Raal, F.J.; Santos, R.D.; Hegele, R.A.; Kuivenhoven, J.A.; Nordestgaard, B.G.; Descamps, O.S.; Steinhagen-Thiessen, E.; et al. Homozygous familial hypercholesterolaemia: New insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 2014, 35, 2146–2157. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Reeskamp, L.F.; Laufs, U.; Banach, M.; Mach, F.; Tokgözoğlu, L.S.; Connolly, D.L.; Gerrits, A.J.; Stroes, E.S.G.; Masana, L.; et al. Combination lipid-lowering therapy as first-line strategy in very high-risk patients. Eur. Heart J. 2022, 43, 830–833. [Google Scholar] [CrossRef]
- Pasta, A.; Cremonini, A.L.; Formisano, E.; Fresa, R.; Bertolini, S.; Pisciotta, L. Long term follow-up of genetically confirmed patients with familial hypercholesterolemia treated with first and second-generation statins and then with PCSK9 monoclonal antibodies. Atherosclerosis 2020, 308, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Vavlukis, M.; Vavlukis, A. Statins Alone or in Combination with Ezetimibe or PCSK9 Inhibitors in Atherosclerotic Cardiovascular Disease Protection. In Lipid Peroxidation Research; IntechOpen: London, UK, 2019. [Google Scholar]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [Green Version]
- Agarwala, A.; Quispe, R.; Goldberg, A.C.; Michos, E.D. Bempedoic Acid for Heterozygous Familial Hypercholesterolemia: From Bench to Bedside. Drug Des. Dev. Ther. 2021, 15, 1955–1963. [Google Scholar] [CrossRef]
- Cuchel, M.; Meagher, E.A.; Theron, H.D.T.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 2013, 381, 40–46. [Google Scholar] [CrossRef] [Green Version]
- German, C.A.; Shapiro, M.D. Small Interfering RNA Therapeutic Inclisiran: A New Approach to Targeting PCSK9. BioDrugs 2020, 34, 1–9. [Google Scholar] [CrossRef]
- Wang, Y.; Gusarova, V.; Banfi, S.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion. J. Lipid Res. 2015, 56, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S.; Burgess, L.J.; Ebenbichler, C.F.; Baum, S.J.; Stroes, E.S.; Ali, S.; Khilla, N.; Hamlin, R.; Pordy, R.; Dong, Y.; et al. Evinacumab in Patients with Refractory Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Ouguerram, K.; Chetiveaux, M.; Zair, Y.; Costet, P.; Abifadel, M.; Varret, M.; Boileau, C.; Magot, T.; Krempf, M. Apolipoprotein B100 Metabolism in Autosomal-Dominant Hypercholesterolemia Related to Mutations in PCSK9. Arter. Thromb. Vasc. Biol. 2004, 24, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidah, N.G.; Awan, Z.; Chrétien, M.; Mbikay, M. PCSK9. Circ. Res. 2014, 114, 1022–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, K.N.; Breslow, J.L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 7100–7105. [Google Scholar] [CrossRef] [Green Version]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations inPCSK9, Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Raal, F.J.; Hovingh, G.K.; Blom, D.; Santos, R.D.; Harada-Shiba, M.; Bruckert, E.; Couture, P.; Soran, H.; Watts, G.F.; Kurtz, C.; et al. Long-term treatment with evolocumab added to conventional drug therapy, with or without apheresis, in patients with homozygous familial hypercholesterolaemia: An interim subset analysis of the open-label TAUSSIG study. Lancet Diabetes Endocrinol. 2017, 5, 280–290. [Google Scholar] [CrossRef]
- Ito, M.K.; Santos, R.D. PCSK9 Inhibition with Monoclonal Antibodies: Modern Management of Hypercholesterolemia. J. Clin. Pharmacol. 2017, 57, 7–32. [Google Scholar] [CrossRef] [Green Version]
- Hovingh, G.K.; Lepor, N.E.; Kallend, D.; Stoekenbroek, R.M.; Wijngaard, P.L.J.; Raal, F.J. Inclisiran Durably Lowers Low-Density Lipoprotein Cholesterol and Proprotein Convertase Subtilisin/Kexin Type 9 Expression in Homozygous Familial Hypercholesterolemia: The ORION-2 Pilot Study. Circulation 2020, 141, 1829–1831. [Google Scholar] [CrossRef]
- Abbasi, J. Highlights from the American Heart Association’s Scientific Sessions-ApoB as a Risk Marker, an Oral PCSK9 Inhibitor, and Aspirin and Dementia. JAMA 2022, 327, 310–312. [Google Scholar] [CrossRef]
- Gennemark, P.; Walter, K.; Clemmensen, N.; Rekić, D.; Nilsson, C.A.; Knöchel, J.; Hölttä, M.; Wernevik, L.; Rosengren, B.; Kakol-Palm, D.; et al. An oral antisense oligonucleotide for PCSK9 inhibition. Sci. Transl. Med. 2021, 13, eabe9117. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.D.; Watts, G.F. Familial hypercholesterolaemia: PCSK9 inhibitors are coming. Lancet 2015, 385, 307–310. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, L.-Y.; Cheng, X.-S. Novel Approaches for the Treatment of Familial Hypercholesterolemia: Current Status and Future Challenges. J. Atheroscler. Thromb. 2018, 25, 665–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Laufs, U.; Banach, M.; Mancini, G.B.J.; Gaudet, D.; Bloedon, L.T.; Sterling, L.R.; Kelly, S.; Stroes, E.S.G. Efficacy and Safety of Bempedoic Acid in Patients with Hypercholesterolemia and Statin Intolerance. J. Am. Heart Assoc. 2019, 8, e011662. [Google Scholar] [CrossRef] [Green Version]
- Lim, G.B. ANGPTL3: A therapeutic target for atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 381. [Google Scholar] [CrossRef]
- Bergmark, B.A.; Marston, N.A.; Bramson, C.R.; Curto, M.; Ramos, V.; Jevne, A.; Kuder, J.F.; Park, J.-G.; Murphy, S.A.; Verma, S.; et al. Effect of Vupanorsen on Non–High-Density Lipoprotein Cholesterol Levels in Statin-Treated Patients with Elevated Cholesterol: TRANSLATE-TIMI 70. Circulation 2022, 145, 1377–1386. [Google Scholar] [CrossRef]
- Ben-Omran, T.; Masana, L.; Kolovou, G.; Ariceta, G.; Nóvoa, F.J.; Lund, A.M.; Bogsrud, M.P.; Araujo, M.; Hussein, O.; Ibarretxe, D.; et al. Real-World Outcomes with Lomitapide Use in Paediatric Patients with Homozygous Familial Hypercholesterolaemia. Adv. Ther. 2019, 36, 1786–1811. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Xu, Y.; Zhang, X.; Ding, C.; Huang, R.; Zhang, Z.; Lv, J.; Xie, X.; Chen, Y.; Li, Y.; et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 2015, 6, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.-T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9–Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef]
- Ajufo, E.; Cuchel, M. Recent Developments in Gene Therapy for Homozygous Familial Hypercholesterolemia. Curr. Atheroscler. Rep. 2016, 18, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Muthuramu, I.; Somanathan, S.; Zhang, H.; Bell, P.; He, Z.; Yu, H.; Zhu, Y.; Tretiakova, A.P.; Wilson, J.M. Developing a second-generation clinical candidate AAV vector for gene therapy of familial hypercholesterolemia. Mol. Ther. Methods Clin. Dev. 2021, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Genetics | 1-Monogenic defects implicated in higher atherosclerotic risk in comparison with hypercholesterolemia of other etiologies [1]. 2-LDL-C concentrations depend not only on defects on canonical genes but also on polygenic effects [2]. |
| Atherosclerosis risk | 1-Risk in heterozygous FH is heterogenous and depends not only on LDL-C but also other biomarkers, genes and subclinical atherosclerosis [3,4,5,6]. 2-Specific FH risk scores [7,8] and coronary atherosclerosis imaging ae useful in risk stratification [4,6]. 3-Risk of homozygous FH is very high, but therapies (drugs and apheresis) have changed the natural history of disease, and with reduction in CHD but persistence of aortic valve disease [9,10,11]. |
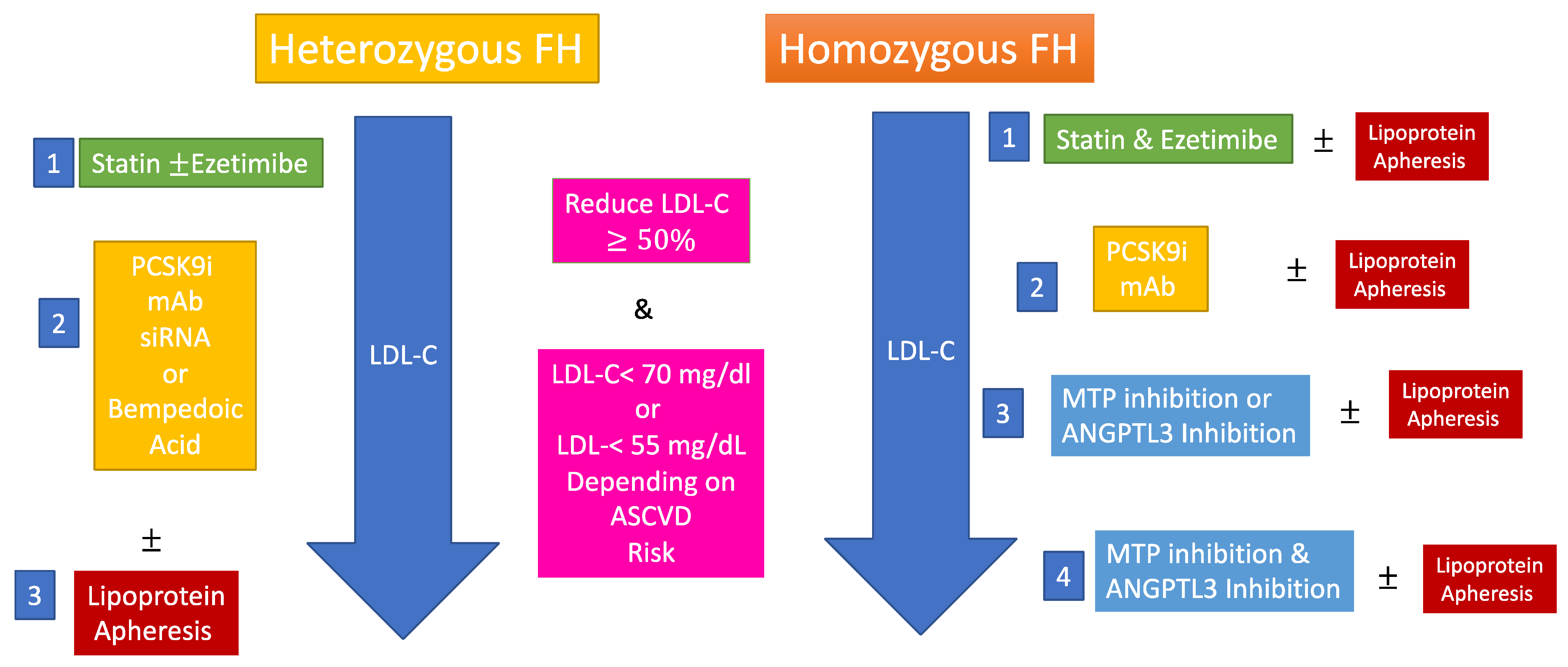
| Therapies | 1-Statin therapy reduces ASCVD risk in FH [12,13] 2-PCSK9 inhibitors have changed the way we treat heterozygous FH but should be used in those at highest risk when not available for all [14]. 3-Combination of statins, ezetimibe, PCSK9 inhibitors, MTP inhibitors and anti ANGPTL3 antibodies may normalize LDL-C in homozygous FH [15,16]. 4-Genetic therapies are being developed for FH and may bring more definitive LDL-C lowering. |
| Compound | Target | Mechanism of Action | Efficacy in Heterozygous FH (LDL-C Reduction) | Efficacy in Homozygous FH (LDL-C Reduction) | |
|---|---|---|---|---|---|
| Statins | Small molecule | HMG-CoA-reductase | Reduces cholesterol synthesis and VLDL production. Increases hepatic LDLR expression [47]. | 30–50% [57]. | 10–25% [53]. |
| Ezetimibe | Small molecule | NPC1L1 | Reduces intestinal cholesterol absorption and increases hepatic LDLR expression [47]. | 10–15% [57]. | 10–15% [53]. |
| Bempedoic acid | Small molecule | ACL | Reduces cholesterol synthesis and VLDL production. Increases LDLR expression [58]. | 16.5% in a pooled group of FH and other hypercholesterolemia patients [58]. | N.A. |
| Lomitapide * | Small molecule | MTP | Reduces VLDL synthesis [47]. | N.A. | 33–50% depending on the dose [15,45,59]. |
| Alirocumab &Evolocumab | Monoclonal antibody | Circulating PCSK9 | Reduces LDLR degradation [47]. | 50–60% [3,33] adults and 35–38% pediatric patients for evolocumab [36,37]. | 20–34% (depends on LDLR variant 0–50%) [33,42]. |
| Inclisiran | Small-interfering RNAs | Hepatic PCSK9 synthesis | Reduces LDLR degradation [60]. | 44.3% reduction [35]. | Study ongoing. |
| Evinacumab * | Monoclonal antibody | Circulating ANGPTL3 | Possibly increases the removal of VLDL and IDL particles by LDLR independent pathways [61]. | 38.5–56% reduction depending on dose regimen and patient [62]. | 49% [16]. |
| Lipoprotein apheresis | Device | Circulating LDL, Lp(a) and VLDL particles | Reduces pro-atherogenic apoB-100-containing lipoproteins LDL, Lp(a), and VLDL as well as pro-inflammatory biomarkers [43]. | 60–80% [3]. | 60–80% [3]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alnouri, F.; Santos, R.D. New Trends and Therapies for Familial Hypercholesterolemia. J. Clin. Med. 2022, 11, 6638. https://doi.org/10.3390/jcm11226638
Alnouri F, Santos RD. New Trends and Therapies for Familial Hypercholesterolemia. Journal of Clinical Medicine. 2022; 11(22):6638. https://doi.org/10.3390/jcm11226638
Chicago/Turabian StyleAlnouri, Fahad, and Raul D. Santos. 2022. "New Trends and Therapies for Familial Hypercholesterolemia" Journal of Clinical Medicine 11, no. 22: 6638. https://doi.org/10.3390/jcm11226638
APA StyleAlnouri, F., & Santos, R. D. (2022). New Trends and Therapies for Familial Hypercholesterolemia. Journal of Clinical Medicine, 11(22), 6638. https://doi.org/10.3390/jcm11226638