
Insights into the Role of Neutrophils and Neutrophil Extracellular Traps in Causing Cardiovascular Complications in Patients with COVID-19: A Systematic Review


Abstract
:1. Introduction
2. Search Method and Systematic Literature Review
3. Results
3.1. Description of the Included Studies and of the Population
3.2. Evidence from Neutrophil Deployment: Target Organs and Mechanism of Action
3.2.1. COVID-19 and Inflammation
3.2.2. Neutrophils Activation: Crucial in SARS-CoV-2 Cardiac Infection
3.2.3. Neutrophils Extracellular Traps in COVID-19: The Hypothesis Takes Shape toward a Defined Role
4. Insights into the Role of Neutrophil Extracellular Traps and Their Interference in the Heart Inflammation Process from SARS-CoV-2 Infection
5. Comment: Myocardial Injury and Mortality in Patients with COVID-19
6. Future Direction
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE1 | angiotensin I-converting enzyme |
| ACE2 | Angiotensin-Converting Enzyme 2 |
| ACEi | ACE–inhibitors |
| aPL | antiphospholipid |
| aPS/PT Ab | anti-phosphatidylserine/prothrombin autoantibodies |
| APS | antiphospholipid syndrome |
| ARDS | acute respiratory distress syndrome |
| AT1R | Angiotensin Type 1 Receptor |
| C | complement |
| CCL | chemokine ligand |
| COPD | Chronic Obstructive Pulmonary Disease |
| COVID-19 | Coronavirus disease-2019 |
| CRP | C-reactive protein |
| CXCL | chemokine ligand |
| CXCL8 | Interleukine 8 |
| CVD | cardiovascular disease |
| DAD | diffuse alveolar damage |
| DIC | disseminated intravascular coagulation |
| ECM | extracellular matrix |
| FDP | fibrinogen derived peptides |
| G-CSF | granulocytes colony-stimulating factor |
| GF | grow factor |
| GM-CSF | granulocyte-macrophage colony stimulating factor |
| HLA-DR | human leucocyte antigen- related D |
| ICAM-1 | intercellular adhesion molecule 1 |
| ICU | intensive care unit |
| IFN | interféron |
| IL | interleukine |
| IP-10 | interferon-gamma-induced protein |
| IRF | interferon regulatory factors |
| ISG-15 | interferon stimulated gene 15 |
| LDG | low-density granulocytes |
| mAb | monoclonal antibody |
| MASP2 | mannose-binding protein associated serine protease 2 |
| MAS | macrophage activation syndrome. |
| MCP-1 | monocyte chemoattractant protein-1 |
| M-CSF | macrophage colony-stimulating factor |
| MIP 1 | macrophage inflammatory protein 1 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NAR | neutrophil count to albumin ratio |
| NCD4LR | neutrophil/CD4 + lymphocyte index |
| NCT | negative conversion time |
| NDG | normal density granulocytes |
| NETs | neutrophil extracellular traps. |
| NHBE | human bronchial epithelial cell |
| NLR | neutrophil-to-lymphocyte ratio |
| NLRP3 | NOD-like receptor family, pyrin domain containing 3 |
| PAD | peptidyl arginine deaminase |
| PAI | platelet activator inhibitor |
| PBMC | peripheral blood immune cells |
| PMN | polymorphonuclear |
| RAAS | renin-angiotensin. -aldosterone system |
| RE | response element |
| ROS | reactive oxygen species |
| SARS-CoV-2 | severe acute respiratory syndrome-coronavirus-2 |
| Sirtuin 3 | SIRT3, |
| STEMI | ST elevation myocardial infarction |
| TF | tissue factor |
| TFPI | tissue factor pathway inhibitor |
| TGF beta-2 | transforming grow factor beta-2 |
| Th | T-helper |
| TNF | tumor necrosis factor |
| TRAP | thrombin receptor-activating peptide |
| β2GPI | β2 I glycoprotein |
References
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular implications of fatal outcomes of patients with Coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szekely, Y.; Lichter, Y.; Taieb, P.; Banai, A.; Hochstadt, A.; Merdler, I.; Oz, A.G.; Rothschild, E.; Baruch, G.; Peri, Y.; et al. The spectrum of cardiac manifestations in coronavirus disease 2019 (COVID-19): A systematic echocardiographic study. Circulation 2020, 142, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Lala, A.; Johnson, K.W.; Januzzi, J.L.; Russak, A.J.; Paranjpe, I.; Richter, F.; Zhao, S.; Somani, S.; Van Vleck, T.; Vaid, A.; et al. Mount Sinai Covid Informatics Center. Prevalence and impact of myocardial injury in patients hospitalized with COVID-19 infection. J. Am. Coll. Cardiol. 2020, 76, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Inciardi, R.M.; Lupi, L.; Zaccone, G.; Italia, L.; Raffo, M.; Tomasoni, D.; Cani, D.S.; Cerini, M.; Farina, D.; Gavazzi, E.; et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 819–824. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef]
- Escher, F.; Pietsch, H.; Aleshcheva, G.; Bock, T.; Baumeier, C.; Elsaesser, A.; Wenzel, P.; Hamm, C.; Westenfeld, R.; Schultheiss, M.; et al. Detection of viral SARS-CoV-2 genomes and histopathological changes in endomyocardial biopsies. ESC Heart Fail. 2020, 7, 2440–2447. [Google Scholar] [CrossRef]
- Hartikainen, T.S.; Sörensen, N.A.; Haller, P.M.; Goßling, A.; Lehmacher, J.; Zeller, T.; Blankenberg, S.; Westermann, D.; Neumann, J.T. Clinical application of the 4th Universal Definition of Myocardial Infarction. Eur. Heart J. 2020, 41, 2209–2216. [Google Scholar] [CrossRef]
- Bonow, R.O.; Fonarow, G.C.; O’Gara, P.T.; Yancy, C.W. Association of coronavirus disease 2019 (COVID-19) with myocardial injury and mortality. JAMA Cardiol. 2020, 5, 751–753. [Google Scholar] [CrossRef] [Green Version]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jiménez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.D.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; Roch, B. Neutrophil extracellular traps and by-products play a key role in COVID-19: Pathogenesis, risk factors, and therapy. J. Clin. Med. 2020, 9, 2942. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.-P.; et al. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Blasco, A.; Coronado, M.J.; Hernández-Terciado, F.H.; Martín, P.; Royuela, A.; Ramil, E.; García, D.; Goicolea, J.; Del Trigo, M.; Ortega, J.; et al. Assessment of Neutrophil Extracellular Traps in Coronary Thrombus of a Case Series of Patients with COVID-19 and Myocardial Infarction. JAMA Cardiol. 2020, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; AlRasheed, M.R.; et al. Pathophysiology of SARS-CoV-2: The Mount Sinai COVID-19 autopsy experience. Mod. Pathol. 2021, 34, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Padera, R.F.; Solomon, I.H.; Kanjilal, S.; Hammer, M.M.; Hornick, J.L.; Sholl, L.M. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Mod. Pathol. 2020, 33, 2104–2114. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. J. Am. Med. Assoc. 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shen, C.; Li, J.; Yuan, J.; Wei, J.; Huang, F.; Wang, F.; Li, G.; Li, Y.; Xing, L.; et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J. Allergy Clin. Immunol. 2020, 146, 119–127.e4. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with, novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Du, X.; Chen, J.; Jin, Y.; Peng, L.; Wang, H.H.X.; Luo, M.; Chen, L.; Zhao, Y. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J. Infect. 2020, 81, e6–e12. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; de Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Burkhard-Koren, N.M.; Haberecker, M.; Maccio, U.; Ruschitzka, F.; Schuepbach, R.A.; Zinkernagel, A.S.; Hardmeier, T.; Varga, Z.; Moch, H. Higher prevalence of pulmonary macrothrombi in SARS-CoV-2 than in influenza A: Autopsy results from ’Spanish flu’ 1918/1919 in Switzerland to Coronavirus disease 2019. J. Pathol. Clin. Res. 2021, 7, 135–143. [Google Scholar] [CrossRef]
- Sang, C.J., 3rd; Burkett, A.; Heindl, B.; Litovsky, S.H.; Prabhu, S.D.; Benson, P.V.; Rajapreyar, I. Cardiac pathology in COVID-19: A single center autopsy experience. Cardiovasc. Pathol. 2021, 54, 107370. [Google Scholar] [CrossRef]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 2021, 595, 114–119. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with coronavirus, (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef]
- Wang, J.; Li, Q.; Yin, Y.; Zhang, Y.; Cao, Y.; Lin, X.; Huang, L.; Hoffmann, D.; Lu, M.; Qiu, Y. Excessive neutrophils and neutrophil extracellular traps in COVID-19. Front. Immunol. 2020, 11, 2063. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Xie, Y.; Bowe, B. High-dimensional characterization of post-acute sequelae of COVID-19. Nature 2021, 594, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Bowe, B.; Maddukuri, G.; Al-Aly, Z. Comparative evaluation of clinical manifestations and risk of death in patients admitted to hospital with COVID-19 and seasonal influenza: Cohort study. BMJ 2020, 371, m4677. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.; Campia, U.; Hurwitz, S.; Snyder, J.E.; Rizzo, S.M.; Pfeferman, M.B.; Morrison, R.B.; Leiva, O.; Fanikos, J.; Nauffal, V.; et al. Registry of arterial and venous thromboembolic complications in patients with COVID-19. J. Am. Coll. Cardiol. 2020, 76, 2060–2072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, W.; Jiang, W.; Xiao, M.; Li, Y.; Tang, N.; Liu, Z.; Yan, X.; Zhao, Y.; Li, T.; et al. Profile of natural anticoagulant, coagulant factor and anti-phospholipid antibody in critically ill COVID-19 patients. J. Thromb. Thrombolysis 2020, 50, 580–586. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Xiang, P.; Pu, L.; Xiong, H.; Li, C.; Zhang, M.; Tan, J.; Xu, Y.; Song, R.; et al. Neutro-philto-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J. Transl. Med. 2020, 18, 206. [Google Scholar] [CrossRef]
- Fu, J.; Kong, J.; Wang, W.; Wu, M.; Yao, L.; Wang, Z.; Jin, J.; Wu, D.; Yu, X. The clinical implication of dynamic neutrophil to lymphocyte ratio and D-dimer in COVID-19: A retroT, s.s.i.S.C.he clinical implication of dynamic neutrophil to lymphocyte ratio and D-dimer in COVID-19: A retrospective study in Suzhou China. Thromb. Res. 2020, 192, 3–8. [Google Scholar] [CrossRef]
- Webb, B.J.; Peltan, I.D.; Jensen, P.; Hoda, D.; Hunter, B.; Silver, A.; Starr, N.; Buckel, W.; Grisel, N.; Hummel, E.; et al. Clinical criteria for COVID-19-associated hyperinflammatory syndrome: A cohort study. Lancet Rheumatol. 2020, 2, e754–e763. [Google Scholar] [CrossRef]
- Ye, W.; Chen, G.; Li, X.; Lan, X.; Ji, C.; Hou, M.; Zhang, D.; Zeng, G.; Wang, Y.; Xu, C.; et al. Dynamic changes of D-dimer and neutrophil-lymphocyte count ratio as prognostic biomarkers in COVID-19. Respir. Res. 2020, 21, 169. [Google Scholar] [CrossRef]
- Tatum, D.; Taghavi, S.; Houghton, A.; Stover, J.; Toraih, E.; Duchesne, J. Neutrophil-to-Lymphocyte ratio and outcomes in Louisiana COVID-19 Patients. Shock 2020, 54, 652–658. [Google Scholar] [CrossRef]
- Yang, A.P.; Liu, J.P.; Tao, W.Q.; Li, H.M. The diagnostic and predictive role of N.L.R., d-NLR and PLR in COVID-19 patients. Int. Immunopharmacol. 2020, 84, 106504. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.; Mo, P.; Liu, J.; Wang, H.; Wang, F.; Zhao, Q. Neutrophil to CD4+ lymphocyte ratio as a potential biomarker in predicting virus negative conversion time in COVID-19. Int. Immunopharmacol. 2020, 85, 106683. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult in patients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a large series of COVID-19 cases from Northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Qu, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. China Medical Treatment Expert Group for COVID-19. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- COVIDSurg Collaborative; GlobalSurg Collaborative. SARS-CoV-2 infection and venous thromboembolism after surgery: An international prospective cohort study. Anaesthesia 2022, 77, 28–39. [Google Scholar]
- COVIDSurg Collaborative; GlobalSurg Collaborative. Effects of pre-operative isolation on postoperative pulmonary complications after elective surgery: An international prospective cohort study. Anaesthesia 2021, 76, 1454–1464. [Google Scholar]
- COVIDSurg Collaborative; GlobalSurg Collaborative. SARS-CoV-2 vaccination modelling for safe surgery to save lives: Data from an international prospective cohort study. Br. J. Surg. 2021, 108, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- COVIDSurg Collaborative; GlobalSurg Collaborative. Timing of surgery following SARS-CoV-2 infection: An international prospective cohort study. Anaesthesia 2021, 76, 748–758. [Google Scholar]
- Xie, Y.; Xu, E.; Bowe, B.; Al-Aly, Z. Long-term cardiovascular outcomes of COVID-19. Nat. Med. 2022, 28, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- García, L.F. Immune response, inflammation, and the clinical spectrum of COVID-19. Front. Immunol. 2020, 11, 1441. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; Chan, J.F.; Wang, Y.; Yuen, T.T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: An ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 71, 1400–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S.; Dandekar, A.A. Immunopathogenesis of coronavirus infections: Implications for SARS. Nat. Rev. Immunol. 2005, 5, 917–927. [Google Scholar]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, M.; Pichlmair, A.; Martínez-Sobrido, L.; Cros, J.; García-Sastre, A.; Haller, O.; Weber, F. Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 2005, 79, 2079–2086. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, A.E. Chemokine receptors: Multifaceted therapeutic targets. Nat. Rev. Immunol. 2002, 2, 106–115. [Google Scholar] [CrossRef]
- Vaninov, N. In the eye of the COVID-19 cytokine storm. Nat. Rev. Immunol. 2020, 20, 27. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 inflammasome in severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- van den Berg, D.F.; Te Velde, A.A. Severe COVID-19: NLRP3 inflammasome dysregulated. Front. Immunol. 2020, 11, 1580. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.L.; Jang, J.W.; Lee, S.W.; Yoo, S.H.; Kwon, J.H.; Nam, S.W.; Bae, S.H.; Choi, J.Y.; Han, N.I.; Yoon, S.K. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci. Rep. 2019, 9, 3260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodarzi, P.; Mahdavi, F.; Mirzaei, R.; Hasanvand, H.; Sholeh, M.; Zamani, F.; Sohrabi, M.; Tabibzadeh, A.; Jeda, A.S.; Niya, M.H.K.; et al. Coronavirus disease 2019 (COVID-19): Immunological ap-proaches and emerging pharmacologic treatments. Int. Immunopharmacol. 2020, 88, 106885. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, C.S.; Miller, S.E.; Martines, R.B.; Bullock, H.A.; Zaki, S.R. Electron microscopy of SARS-CoV-2: A challenging task. Lancet 2020, 395, e99. [Google Scholar] [CrossRef]
- Tan, M.; Liu, Y.; Zhou, R.; Deng, X.; Li, F.; Liang, K.; Shi, Y. Immunopathological characteristics of coronavirus disease, cases in Guangzhou China. Immunology 2020, 160, 261–268. [Google Scholar] [CrossRef]
- Nappi, F.; Giacinto, O.; Ellouze, O.; Nenna, A.; Avtaar Singh, S.S.; Chello, M.; Bouzguenda, A.; Copie, X. Association between COVID-19 Diagnosis and Coronary Artery Thrombosis: A Narrative Review. Biomedicines 2022, 10, 702. [Google Scholar] [CrossRef]
- Soehnlein, O.; Steffens, S.; Hidalgo, A.; Weber, C. Neutrophils as protagonists and targets in chronic inflammation. Nat. Rev. Immunol. 2017, 17, 248–261. [Google Scholar] [CrossRef]
- Tomar, B.; Anders, H.J.; Desai, J.; Mulay, S.R. Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID-19. Cells 2020, 9, 1383. [Google Scholar] [CrossRef]
- Galani, I.E.; Andreakos, E. Neutrophils in viral infections: Current concepts and caveats. J. Leukoc. Biol. 2015, 98, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Naumenko, V.; Turk, M.; Jenne, C.N.; Kim, S.J. Neutrophils in viral infection. Cell Tissue Res. 2018, 371, 505–516. [Google Scholar] [CrossRef]
- Camp, J.V.; Jonsson, C.B. A role for neutrophils in viral respiratory disease. Front. Immunol. 2017, 8, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmat, N.; Derakhshani, A.; Bannazadeh Baghi, H.; Silvestris, N.; Baradaran, B.; De Summa, S. Neutrophils, crucial, or harmful immune cells involved in coronavirus infection: A bioinformatics study. Front. Genet. 2020, 11, 641. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zheng, H.; Yin, Y.F.; Hu, Q.Y.; Teng, J.L.; Sun, Y.; Liu, H.L.; Cheng, X.B.; Ye, J.N.; Su, Y.T.; et al. Antiphosphatidylserine/prothrombin antibodies (aPS/PT) as potential diagnostic markers and risk predictors of venous thrombosis and obstetric complications in antiphospholipid syndrome. Clin. Chem. Lab. Med. 2018, 56, 614–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Y.; Shi, H.; Li, C.; Knight, J.S. Antiphospholipid syndrome: A clinical perspective. Chin. Med. J. 2020, 133, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Wei, X.; Qin, S.; Liu, X.; Jiang, Y.; Chen, Y.; Chen, Y.; Lu, H.; Qian, J.; Wang, Z.; et al. Continuous tracking of COVID-19 patients’ immune status. Int. Immunopharmacol. 2020, 89 (Pt A), 107034. [Google Scholar] [CrossRef]
- Ponti, G.; Maccaferri, M.; Ruini, C.; Tomasi, A.; Ozben, T. Biomarkers associated with COVID-19 disease progression. Crit. Rev. Clin. Lab. Sci. 2020, 57, 389–399. [Google Scholar] [CrossRef]
- Azab, B.; Camacho-Rivera, M.; Taioli, E. Average values and racial differences of neutrophil lymphocyte ratio among a nationally representative sample of United States subjects. PLoS ONE 2014, 9, e112361. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Zhang, S.; Hu, H.; Liu, T.; Huang, J. The role of neutrophil-lymphocyte ratio and lymphocyte–monocyte ratio in the prognosis of type 2 diabetics with COVID-19. Scott. Med. J. 2020, 65, 154–160. [Google Scholar] [CrossRef]
- Yang, P.; Wang, N.; Wang, J.; Luo, A.; Gao, F.; Tu, Y. Admission fasting plasma glucose is an independent risk factor for 28-day mortality in patients with COVID-19. J. Med. Virol. 2021, 93, 2168–2176. [Google Scholar] [CrossRef]
- Varim, C.; Yaylaci, S.; Demirci, T.; Kaya, T.; Nalbant, A.; Dheir, H.; Senocak, D.; Kurt, R.; Cengiz, H.; Karacaer, C. Neutrophil count to albumin ratio as a new predictor of mortality in patients with COVID-19 ınfection. Rev. Assoc. Med. Bras. 2020, 66 (Suppl. 2), 77–81. [Google Scholar] [CrossRef]
- Horton, R. Offline: COVID-19—Bewilderment and candour. Lancet 2020, 395, 1178. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef] [PubMed]
- Reusch, N.; De Domenico, E.; Bonaguro, L.; Schulte-Schrepping, J.; Baßler, K.; Schultze, J.L.; Aschenbrenner, A.C.A. Neutrophils in COVID-19. Front. Immunol. 2021, 12, 652470. [Google Scholar] [CrossRef]
- Parackova, Z.; Zentsova, I.; Bloomfield, M.; Vrabcova, P.; Smetanova, J.; Klocperk, A.; Mesežnikov, G.; Casas Mendez, L.F.; Vymazal, T.; Sediva, A. Disharmonic inflammatory signatures in COVID-19 Augmented neutrophils’ but impaired monocytes’ and dendritic cells’ responsiveness. Cells 2020, 9, 2206. [Google Scholar] [CrossRef]
- Cicco, S.; Cicco, G.; Racanelli, V.; Vacca, A. Neutrophil Extracellular Traps (NETs) and Damage-Associated Molecular Patterns (DAMPs): Two potential targets for COVID-19 treatment. Mediat. Inflamm. 2020, 2020, 7527953. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Demers, M.; Wagner, D.D. NETosis: A new factor in tumour progression and cancer associated thrombosis. Semin. Thromb. Hemost. 2014, 40, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Sangaletti, S.; Tripodo, C.; Chiodoni, C.; Guarnotta, C.; Cappetti, B.; Casalini, P.; Piconese, S.; Parenza, M.; Guiducci, C.; Vitali, C.; et al. Neutrophil Extracellular Traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 2012, 120, 3007–3018. [Google Scholar] [CrossRef] [Green Version]
- Mozzini, C.; Garbin, U.; Fratta Pasini, A.M.; Cominacini, L. An exploratory look at NETosis in atherosclerosis. Intern. Emerg. Med. 2017, 12, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Almyroudis, N.G.; Grimm, M.J.; Davidson, B.A.; Röhm, M.; Urban, C.F.; Segal, B.H. NETosis and NADPH oxidase: At the intersection of host defence, inflammation, and injury. Front. Immunol. 2013, 4, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoiber, W.; Obermayer, A.; Steinbacher, P.; Krautgartner, W.D. The role of reactive oxygen species (ROS) in the formation of extracellular traps in humans. Biomolecules 2015, 5, 702–723. [Google Scholar] [CrossRef] [Green Version]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Schönrich, G.; Raftery, M.J. Neutrophil Extracellular Traps go viral. Front. Immunol. 2016, 7, 366. [Google Scholar] [CrossRef] [Green Version]
- Hiroki, C.H.; Toller-Kawahisa, J.E.; Fumagalli, M.J.; Colon, D.F.; Figueiredo, L.T.M.; Fonseca, B.A.L.D.; Franca, R.F.O.; Cunha, F.Q. Neutrophil Extracellular Traps effectively control acute Chikungunya virus infection. Front. Immunol. 2020, 10, 3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraro, S.P.; de Souza, G.F.; Gallo, S.W.; de Silva, B.K.; de Oliveira, S.D.; Vinolo, M.A.R.; Saraiva, E.M.; Porto, B.N. Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci. Rep. 2018, 8, 14166. [Google Scholar] [CrossRef]
- Mikacenic, C.; Moore, R.; Dmyterko, V.; West, T.E.; Altemeier, W.A.; Liles, W.C.; Lood, C. Neutrophil Extracellular Traps are increased in the alveolar spaces of patients with ventilator-associated pneumonia. Crit. Care 2018, 22, 358. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.J.M.; Leong, J.Y.; Lee, J.H.; Albani, S.; Yeo, J.G. Insights into the immune-pathogenesis of acute respiratory distress syndrome. Ann. Transl. Med. 2019, 7, 504. [Google Scholar] [CrossRef]
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, W.D.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir. Res. 2015, 16, 59. [Google Scholar] [CrossRef] [Green Version]
- Baden, L.R.; Rubin, E.J. COVID-19: The search of effective therapy. N. Engl. J. Med. 2020, 382, 1851–1852. [Google Scholar] [CrossRef] [PubMed]
- Madison, J.A.; Duarte-García, A.; Zuo, Y.; Knight, J.S. Treatment of thrombotic antiphospholipid syndrome in adults and children. Curr. Opin. Rheumatol. 2020, 32, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: A newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and antiphospholipid antibodies in patients with COVID-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef]
- Siguret, V.; Voicu, S.; Neuwirth, M.; Delrue, M.; Gayat, E.; Stépanian, A.; Mégarbane, B. Are antiphospholipid antibodies associated with thrombotic complications in critically ill COVID-19 patients? Thromb. Res. 2020, 195, 74–76. [Google Scholar] [CrossRef]
- Shi, H.; Zuo, Y.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Woodward, W.; Lezak, S.P.; Lugogo, N.L.; et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J. Leukoc. Biol. 2021, 109, 67–72. [Google Scholar] [CrossRef]
- Blasco, A.; Bellas, C.; Goicolea, L.; Muñiz, A.; Abraira, V.; Royuela, A.; Mingo, S.; Oteo, J.F.; García-Touchard, A.; Goicolea, F.J. Immunohistological analysis of intracoronary thrombus aspirate in STEMI patients: Clinical implications of pathological findings. Rev. Esp. Cardiol. 2017, 70, 170–177. [Google Scholar] [CrossRef]
- Nicin, L.; Abplanalp, W.T.; Mellentin, H.; Kattih, B.; Tombor, L.; John, D.; Schmitto, j.; Heineke, J.; Emrich, F.; Arsalan, M.; et al. Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 2020, 41, 1804–1806. [Google Scholar] [CrossRef] [Green Version]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.; Penninger, J.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Zhang, X.; Tan, Y.; Ling, Y.; Lu, G.; Liu, F.; Yi, Z.; Jia, X.; Wu, M.; Shi, B.; Xu, S.; et al. Viral and host factors related to the clinical outcome of COVID-19. Nature 2020, 583, 437–440. [Google Scholar] [CrossRef]
- Nappi, F.; Iervolino, A.; Avtaar Singh, S.S. COVID-19 Pathogenesis: From Molecular Pathway to Vaccine Administration. Biomedicines 2021, 9, 903. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Iervolino, A.; Avtaar Singh, S.S. Thromboembolic Complications of SARS-CoV-2 and Metabolic Derangements: Suggestions from Clinical Practice Evidence to Causative Agents. Metabolites 2021, 11, 341. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F. Incertitude Pathophysiology and Management During the First Phase of the COVID-19 Pandemic. Ann. Thorac. Surg. 2022, 113, 693. [Google Scholar] [CrossRef] [PubMed]
- Madjid, M.; Miller, C.C.; Zarubaev, V.V.; Marinich, I.G.; Kiselev, O.I.; Lobzin, Y.V.; Filippov, A.E.; Casscells, S.W. Influenza epidemics and acute respiratory disease activity are associated with a surge in autopsy-confirmed coronary heart disease death: Results from years of autopsies in 34,892 subjects. Eur. Heart J. 2007, 28, 1205–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, J.L.; Yang, W.; Ito, K.; Matte, T.D.; Shaman, J.; Kinney, P.L. Seasonal influenza infections and cardiovascular disease mortality. JAMA Cardiol. 2016, 1, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, J.C.; Schwartz, K.L.; Campitelli, M.A.; Chung, H.; Crowcroft, N.S.; Karnauchow, T.; Katz, K.; Ko, D.T.; McGeer, A.J.; McNally, D.; et al. Acute myocardial infarction after laboratory-confirmed influenza infection. N. Engl. J. Med. 2018, 378, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Smeeth, L.; Thomas, S.L.; Hall, A.J.; Hubbard, R.; Farrington, P.; Vallance, P. Risk of myocardial infarction and stroke after acute infection or vaccination. N. Engl. J. Med. 2004, 351, 2611–2618. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Jin, Z. An acute respiratory infection runs into the most common noncommunicable epidemic—COVID-19 and cardiovascular diseases. JAMA Cardiol. 2020, 5, 743–744. [Google Scholar] [CrossRef] [Green Version]
- Madjid, M.; Safavi-Naeini, P.; Solomon, S.D.; Vardeny, O. Potential effects of coronaviruses on the cardiovascular system: A review. JAMA Cardiol. 2020, 5, 831–840. [Google Scholar] [CrossRef] [Green Version]
- Moorlag, S.J.C.F.M.; Rodriguez-Rosales, Y.A.; Gillard, J.; Fanucchi, S.; Theunissen, K.; Novakovic, B.; de Bont, C.M.; Negishi, Y.; Fok, E.T.; Kalafati, L.; et al. BCG Vaccination Induces Long-Term Functional Reprogramming of Human Neutrophils. Cell Rep. 2020, 33, 108387. [Google Scholar] [CrossRef]
- Kalafati, L.; Kourtzelis, I.; Schulte-Schrepping, J.; Li, X.; Hatzioannou, A.; Grinenko, T.; Hagag, E.; Sinha, A.; Has, C.; Dietz, S.; et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 2020, 183, 771–785.e12. [Google Scholar] [CrossRef] [PubMed]
- Lajqi, T.; Köstlin-Gille, N.; Hillmer, S.; Braun, M.; Kranig, S.A.; Dietz, S.; Krause, C.; Rühle, J.; Frommhold, D.; Pöschl, J.; et al. Gut Microbiota-Derived Small Extracellular Vesicles Endorse Memory-like Inflammatory Responses in Murine Neutrophils. Biomedicines 2022, 10, 442. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author/Year | Study Period | Total Number | COVID-19 Study Design | Hospitals/Centers | Type |
|---|---|---|---|---|---|
| Shi (2020) [1] JAMA | 20 January 2020 to 10 February 2020 | 416 | Clinical, laboratory, radiological, and treatment | Single Center Wuhan, China | Prospective |
| Guo (2020) [2] JAMA Cardiology | 20 January 2020 to 10 February 2020 | 187 | Clinical laboratory comorbidities, and treatments | Single Center Wuhan, China | Observational |
| Szekely (2020) [3] Circulation | 21 March 2020 to 16 April 2020 | 100 | Echocardiographic | Single Center Israel | Prospective |
| Lala (2020) [4] JACC | 27 February 2020 to 12 April 2020 | 506 | Clinical, laboratory, Echocardiographic | Single Center NYC, NY, USA | Prospective |
| Escher (2020) [7] ESC Heart Fail | 3 February 2020 to 26 March 2020 | 104 | Endomyocardial biopsies | Multicenter Germany | Prospective |
| Lindner (2020) [13] JAMA Cardiology | 8 April 2020 to 18 April 2020 | 39 | Autopsy | Multicenter Germany | Prospective |
| Blasco (2020) [14] JAMA Cardiology | 24 March 2020 to 11 April 2020 | 55 | PCI/Coronary aspirates, NETs | Single Center Spain | Prospective |
| Ackermann (2020) [15] NEJM | 2019 † 2009 †† | 24 | Pulmonary autopsy/Immune profiling | Multicenter Germany/USA | Comparative study |
| Bryce (2021) [16] Mod. Pathol. | 20 March 2020 to 23 June 2020 | 100 | Pulmonary autopsy/Immune profiling | Single Center NYC, NY, USA | Prospective |
| Schaefer (2020) [17] Mod. Pathol. | April 2020 | 7 | Pulmonary autopsy/Immune profiling | Single Center Boston, MA, USA | Observational |
| Varga (2020) [18] Lancet | « « « | 3 | Autopsy/Immune profiling | Multicenter Switzerland/USA | Observational |
| Delorey (2021) [19] Nature | « « « | 17 | Autopsy/Immune profiling | Multicenter USA | Comparative study |
| Wang (2020) [20] JAMA | 1 January 2020 to 28 January 2020 | 138 | Clinical, laboratory, radiological, and treatment | Single Center Wuhan, China | Observational |
| Lucas (2020) [21] Nature | 18 March 2020 to 27 May 2020 | 113 | Immune profiling | Multicenter USA | Observational |
| Yang (2020) [22] J Allergy Clin. Immunol. | « « « | 50 | Immune profiling | Multicenter China | Observational |
| Huang (2020) [23] Lancet | 16 December 2019 to 2 January 2020 | 41 | Immune profiling | Multicenter China | Observational |
| Liu (2020) [24] J. Infect. | 11 January 2020 to 29 January 2020 | 245 | Immune profiling | Multicenter China/UK | Observational |
| Rodriguez (2021) [25] J. Exp. Med. | « « « | 124 | Autopsy/Immune profiling | Multicenter Brasil | Observational |
| Burkhard-Koren (2021) [26] J. Pathol. Clin. Res. | May 1918 to April 1919 2009–2020 Until 2020 | 411 | Autopsy/Immune profiling | Single center Switzerland | Comparative study |
| Sang (2021) [27] Cardiovasc. Pathol. | Until 2021 | 50 | Autopsy/Immune profiling | Single Center Birmingham, AL, USA | Observational |
| Melms (2021) [28] Nature | Until 2021 | 26 | Autopsy/Immune profiling | Multicenter USA | Comparative study |
| Qin (2020) [29] Clin. Infect. Dis. | 10 January 2020 to 12 February 2020 | 452 | Immune profiling | Single Center Wuhan, China | Observational |
| Wilk (2020) [30] Nat. Med. | March–April 2020 | 7 | Immune profiling | Single Center Stanford, CA, USA | Prospective |
| Wang (2020) [31] Front. Immunol. | 23 January 2020 to 15 March 2020 | 55 | Immune profiling/NETs | Multicenter China/Germany | Observational |
| Al-Aly (2021) [32] Nature | Until 2021 | 73,435 | Clinical, laboratory | Single Center Saint Louis, MO, USA | Observational |
| Xie (2020) [33] Br. Med. J. | 1 January 2017 to 31 January 2019 2 January 2020 to 17 June 2020 | 16,317 | Clinical, laboratory | Single Center Saint Louis, MO, USA | Comparative study |
| Piazza (2020) [34] JACC | 13 March 2020 to 3 April 2020 | 1114 | Clinical Thromboembolic Complication | Single Center Boston, MA, USA | Observational |
| Zhang (2020) [35] J. Thromb. Thrombolysis | 23 February 2020 to 3 March 2020 | 12 | Clinical Thromboembolic Complication | Multicenter China | Prospective |
| Liu (2020) [36] J. Transl. Med. | 1 February 2020 to 24 February 2020 | 61 | Immune profiling | Single Center Beijing, China | Prospective |
| Fu (2020) [37] Thromb. Res. | 20 January 2020 to 20 February 2020 | 75 | Immune profiling Thromboembolic Complication | Single Center Suzhou, China | Comparative study |
| Webb (2020) [38] Lancet Rheumatol. | 13 March 2020 to 5 May 2020 | 299 | Immune profiling | Multicenter USA | Observational |
| Ye (2020) [39] Respir. Res. | 1 January 2020 to 16 March 2020 | 349 | Immune profiling Thromboembolic Complication | Multicenter China | Prospective |
| Tatum (2020) [40] Shock | Until 2021 | 125 | Immune profiling | Multicenter USA | Multicenter Prospective Registry |
| Yang (2020) [41] Int. Immunopharmacol. | Until 20 February 2020 | 93 | Immune profiling | Multicenter China | Observational |
| Wang (2020) [42] Int. Immunopharmacol. | 15 January 2020 to 2 March 2020 | 95 | Immune profiling | Single Center Wuhan, China | Observational |
| Zhou (2020) [43] Lancet | 29 December 2019 to 30 January 2020 | 191 | Clinical, laboratory, radiological, and treatment | Multicenter China | Observational |
| Klok (2020) [44] Thromb. Res. | 7 March 2020 to 5 April 2020 | 184 | Thromboembolic Complication | Multicenter Netherlands | Prospective |
| Tang (2020) [45] J. Thromb. Haemost. | 1 January 2020 to 13 February 2020 | 448 | Thromboembolic Complication | Single Center Wuhan, China | Observational |
| Zuo (2020) [46] Sci. Transl. Med. | « « « « | 172 | Immune profiling Thromboembolic Complication/NETs | Multicenter China/USA | Prospective |
| Carsana (2020) [47] Lancet Infect. Dis. | 29 February 2020 to 24 March 2020 | 38 | Autopsy/Immune profiling | Multicenter Italy | Observational |
| Chen (2020) [48] Lancet | 1 January 2020 to 20 January 2020 | 99 | Clinical, laboratory, radiological, and treatment | Multicenter China | Observational |
| Guan (2020) [49] NEJM | 11 December 2019 to 29 January 2020 | 1099 | Clinical, laboratory, radiological, and treatment | Multicenter China | Observational |
| COVIDSurg Collaborative (2022) [50] Anaesthesia | 10 January 2020 to 30 January 2020 | 128,013 | Thromboembolic Complication | Multicenter | Prospective |
| COVIDSurg Collaborative (2021) [51] Anaesthesia | 10 January 2020 to 30 January 2020 | 96,454 | Clinical | Multicenter | Prospective |
| COVIDSurg Collaborative (2021) [52] Br. J. Surg. | 10 January 2020 to 30 January 2020 | 56,589 | Clinical/Vaccine effectiveness | Multicenter | Prospective |
| COVIDSurg Collaborative (2021) [53] Anaesthesia | 10 January 2020 to 30 January 2020 | 140,231 | Clinical | Multicenter | Prospective |
| Xie (2022) [54] Nat. Med. | 1 March 2020 to 15 January 2021 | 153,760 | Clinical | Multicenter USA | Observational |
| Section and Topic | Item # | Checklist Item | Location Where Item Is Reported |
|---|---|---|---|
| TITLE | |||
| Title | 1 | Identify the report as a systematic review. | Title and introduction |
| ABSTRACT | |||
| Abstract | 2 | See the PRISMA 2020 for Abstracts checklist. | Abstract |
| INTRODUCTION | |||
| Rationale | 3 | Describe the rationale for the review in the context of existing knowledge. | Introduction |
| Objectives | 4 | Provide an explicit statement of the objective(s) or question(s) the review addresses. | Introduction |
| METHODS | |||
| Eligibility criteria | 5 | Specify the inclusion and exclusion criteria for the review and how studies were grouped for the syntheses. | Methods |
| Information sources | 6 | Specify all databases, registers, websites, organisations, reference lists and other sources searched or consulted to identify studies. Specify the date when each source was last searched or consulted. | Methods/PRISMA statement |
| Search strategy | 7 | Present the full search strategies for all databases, registers and websites, including any filters and limits used. | Methods |
| Selection process | 8 | Specify the methods used to decide whether a study met the inclusion criteria of the review, including how many reviewers screened each record and each report retrieved, whether they worked independently, and if applicable, details of automation tools used in the process. | Methods |
| Data collection process | 9 | Specify the methods used to collect data from reports, including how many reviewers collected data from each report, whether they worked independently, any processes for obtaining or confirming data from study investigators, and if applicable, details of automation tools used in the process. | Methods |
| Data items | 10a | List and define all outcomes for which data were sought. Specify whether all results that were compatible with each outcome domain in each study were sought (e.g., for all measures, time points, analyses), and if not, the methods used to decide which results to collect. | Methods |
| 10b | List and define all other variables for which data were sought (e.g. participant and intervention characteristics, funding sources). Describe any assumptions made about any missing or unclear information. | Methods | |
| Study risk of bias assessment | 11 | Specify the methods used to assess risk of bias in the included studies, including details of the tool(s) used, how many reviewers assessed each study and whether they worked independently, and if applicable, details of automation tools used in the process. | n/a |
| Effect measures | 12 | Specify for each outcome the effect measure(s) (e.g., risk ratio, mean difference) used in the synthesis or presentation of results. | n/a |
| Synthesis methods | 13a | Describe the processes used to decide which studies were eligible for each synthesis (e.g., tabulating the study intervention characteristics and comparing against the planned groups for each synthesis (item #5)). | Methods |
| 13b | Describe any methods required to prepare the data for presentation or synthesis, such as handling of missing summary statistics, or data conversions. | n/a | |
| 13c | Describe any methods used to tabulate or visually display results of individual studies and syntheses. | Methods | |
| 13d | Describe any methods used to synthesize results and provide a rationale for the choice(s). If meta-analysis was performed, describe the model(s), method(s) to identify the presence and extent of statistical heterogeneity, and software package(s) used. | n/a | |
| 13e | Describe any methods used to explore possible causes of heterogeneity among study results (e.g., subgroup analysis, meta-regression). | n/a | |
| 13f | Describe any sensitivity analyses conducted to assess robustness of the synthesized results. | n/a | |
| Reporting bias assessment | 14 | Describe any methods used to assess risk of bias due to missing results in a synthesis (arising from reporting biases). | n/a |
| Certainty assessment | 15 | Describe any methods used to assess certainty (or confidence) in the body of evidence for an outcome. | n/a |
| RESULTS | |||
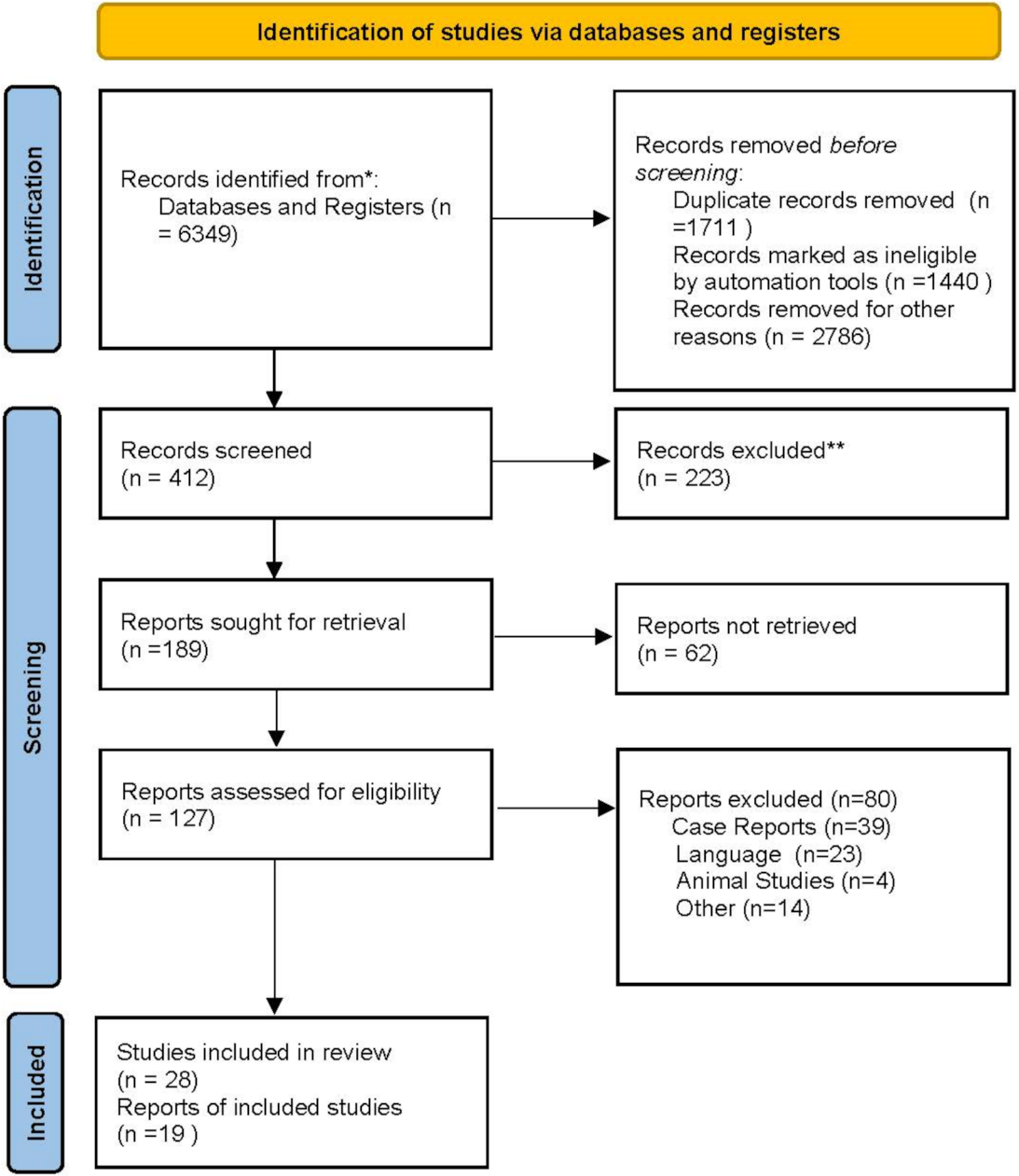
| Study selection | 16a | Describe the results of the search and selection process, from the number of records identified in the search to the number of studies included in the review, ideally using a flow diagram. | Prisma diagram |
| 16b | Cite studies that might appear to meet the inclusion criteria, but which were excluded, and explain why they were excluded. | Prisma diagram | |
| Study characteristics | 17 | Cite each included study and present its characteristics. | Table 1 |
| Risk of bias in studies | 18 | Present assessments of risk of bias for each included study. | n/a |
| Results of individual studies | 19 | For all outcomes, present, for each study: (a) summary statistics for each group (where appropriate) and (b) an effect estimate and its precision (e.g., confidence/credible interval), ideally using structured tables or plots. | n/a |
| Results of syntheses | 20a | For each synthesis, briefly summarise the characteristics and risk of bias among contributing studies. | n/a |
| 20b | Present results of all statistical syntheses conducted. If meta-analysis was carried out, present for each the summary estimate and its precision (e.g., confidence/credible interval) and measures of statistical heterogeneity. If comparing groups, describe the direction of the effect. | Table 1 | |
| 20c | Present results of all investigations of possible causes of heterogeneity among study results. | n/a | |
| 20d | Present results of all sensitivity analyses conducted to assess the robustness of the synthesized results. | n/a | |
| Reporting biases | 21 | Present assessments of risk of bias due to missing results (arising from reporting biases) for each synthesis assessed. | n/a |
| Certainty of evidence | 22 | Present assessments of certainty (or confidence) in the body of evidence for each outcome assessed. | n/a |
| DISCUSSION | |||
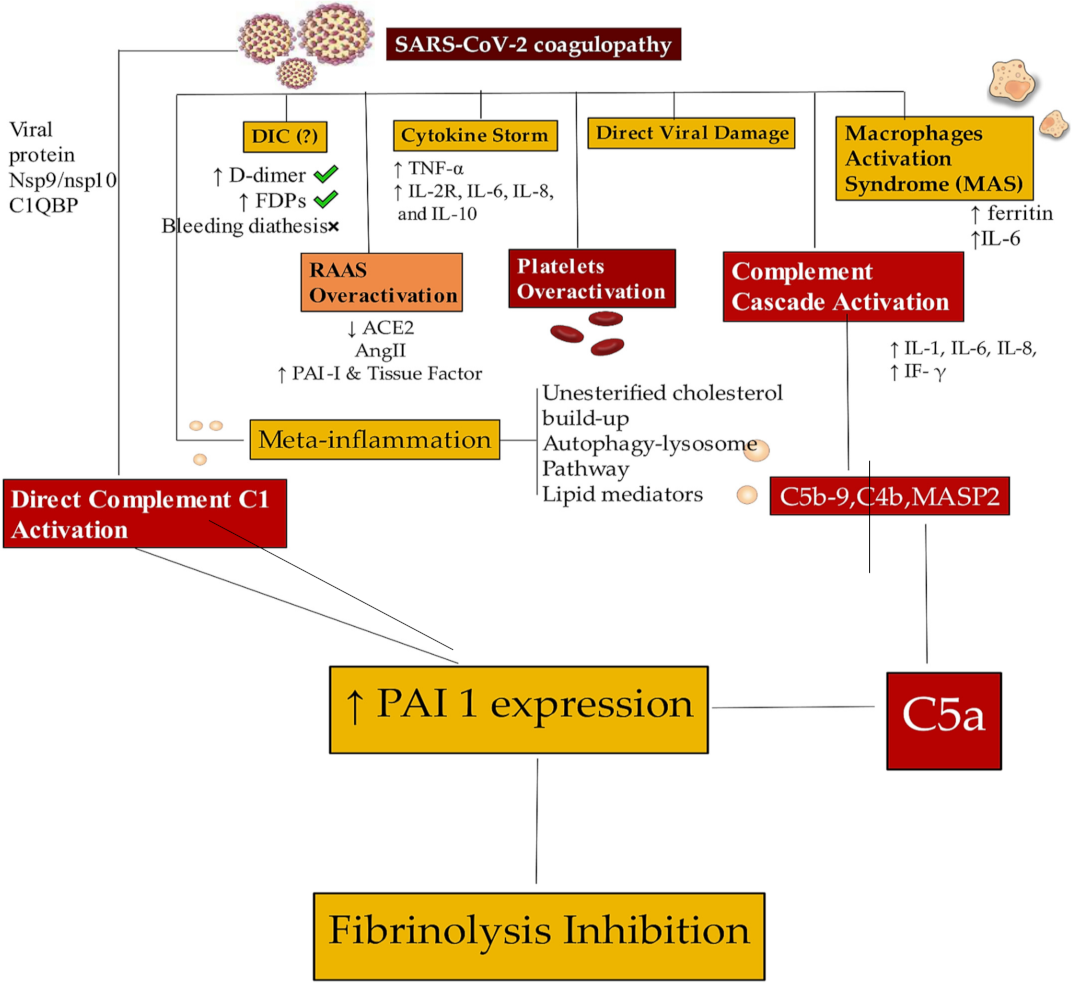
| Discussion | 23a | Provide a general interpretation of the results in the context of other evidence. | 3.2 |
| 23b | Discuss any limitations of the evidence included in the review. | n/a | |
| 23c | Discuss any limitations of the review processes used. | n/a | |
| 23d | Discuss implications of the results for practice, policy, and future research. | 3.2 | |
| OTHER INFORMATION | |||
| Registration and protocol | 24a | Provide registration information for the review, including register name and registration number, or state that the review was not registered. | Methods |
| 24b | Indicate where the review protocol can be accessed, or state that a protocol was not prepared. | Methods | |
| 24c | Describe and explain any amendments to information provided at registration or in the protocol. | n/a | |
| Support | 25 | Describe sources of financial or non-financial support for the review, and the role of the funders or sponsors in the review. | Methods |
| Competing interests | 26 | Declare any competing interests of review authors. | Methods |
| Availability of data, code and other materials | 27 | Report which of the following are publicly available and where they can be found: template data collection forms; data extracted from included studies; data used for all analyses; analytic code; any other materials used in the review. | n/a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nappi, F.; Bellomo, F.; Avtaar Singh, S.S. Insights into the Role of Neutrophils and Neutrophil Extracellular Traps in Causing Cardiovascular Complications in Patients with COVID-19: A Systematic Review. J. Clin. Med. 2022, 11, 2460. https://doi.org/10.3390/jcm11092460
Nappi F, Bellomo F, Avtaar Singh SS. Insights into the Role of Neutrophils and Neutrophil Extracellular Traps in Causing Cardiovascular Complications in Patients with COVID-19: A Systematic Review. Journal of Clinical Medicine. 2022; 11(9):2460. https://doi.org/10.3390/jcm11092460
Chicago/Turabian StyleNappi, Francesco, Francesca Bellomo, and Sanjeet Singh Avtaar Singh. 2022. "Insights into the Role of Neutrophils and Neutrophil Extracellular Traps in Causing Cardiovascular Complications in Patients with COVID-19: A Systematic Review" Journal of Clinical Medicine 11, no. 9: 2460. https://doi.org/10.3390/jcm11092460
APA StyleNappi, F., Bellomo, F., & Avtaar Singh, S. S. (2022). Insights into the Role of Neutrophils and Neutrophil Extracellular Traps in Causing Cardiovascular Complications in Patients with COVID-19: A Systematic Review. Journal of Clinical Medicine, 11(9), 2460. https://doi.org/10.3390/jcm11092460