
Current Advances in Gene Therapies of Genetic Auditory Neuropathy Spectrum Disorder

 ,
, 
 , ,
, , 
Abstract
:1. Introduction
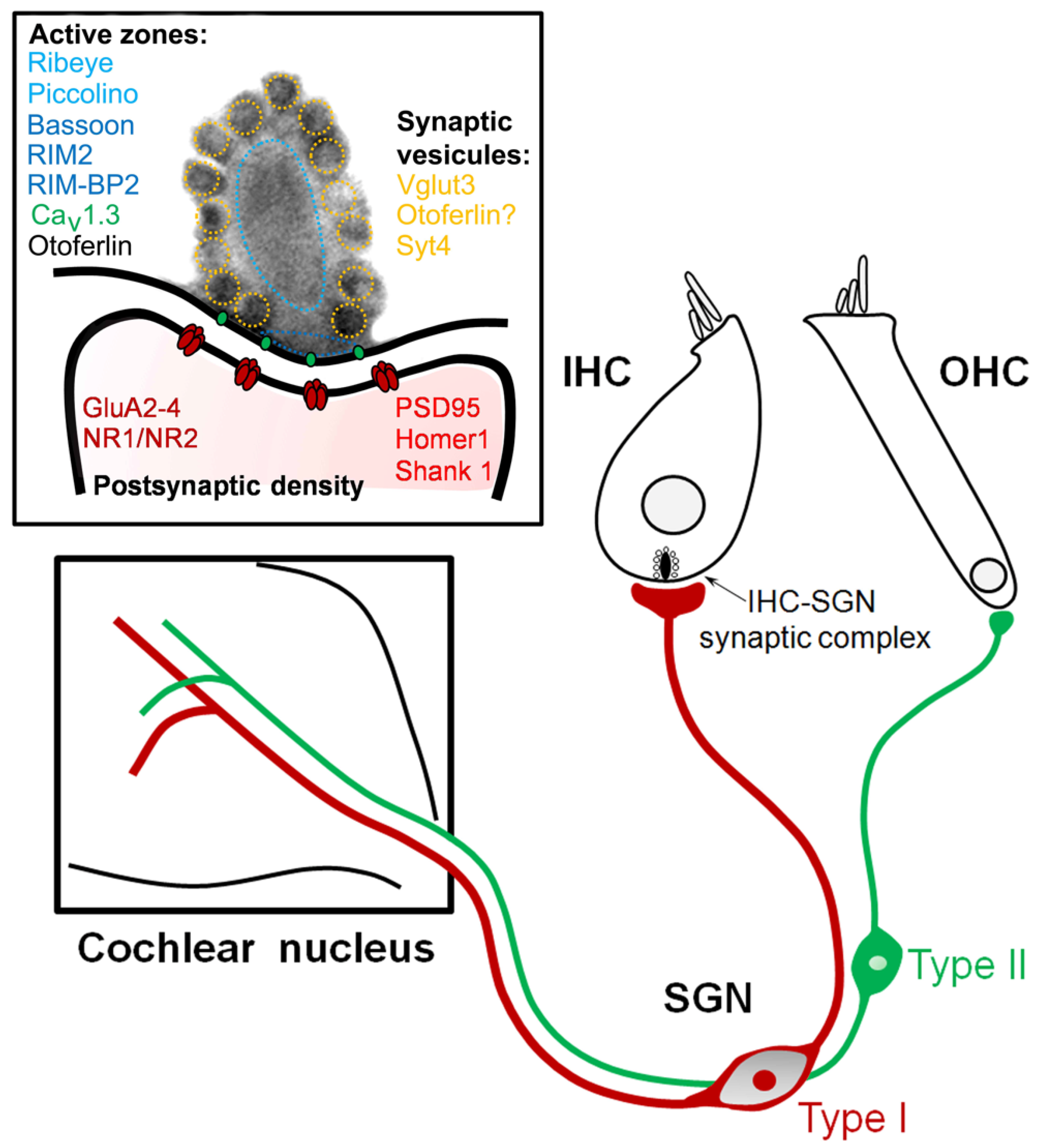
2. Pathogenic Mechanisms of Auditory Neuropathy
2.1. Non-Syndromic Auditory Synaptopathies
2.1.1. Otoferlin-DFNB9
2.1.2. VGLUT3-DFNA25
2.1.3. Cav1.3-SANDD
2.1.4. CABP2-DFNB93
2.1.5. DIAPH3-AUNA1
2.2. Syndromic Auditory Neuropathy
2.2.1. Charcot–Marie–Tooth
2.2.2. Autosomal-Dominant Optic Atrophy
2.2.3. Leber Hereditary Optic Neuropathy
2.2.4. Friedreich’s Ataxia
2.2.5. Mohr–Tranebjaerg Syndrome
3. Gene Therapies for Genetic Synaptopathies and Neuropathies
3.1. Restoration of Neurotransmission in IHC Synapses
3.1.1. DFNB9
3.1.2. DFNA25
3.1.3. DFNB93
3.2. Hearing Restoration in Syndromic Auditory Neuropathy
3.2.1. Charcot–Marie–Tooth
3.2.2. Autosomal-Dominant Optic Atrophy
3.2.3. Leber Hereditary Optic Neuropathy (LHON)

{kind=link}
{kind=link}
{kind=link}
| Diseases | Defective Genes | Therapeutic Strategies | Benefic Effects | Clinical Trials |
|---|---|---|---|---|
| DFNB9 | OTOF | AAV-synaptotagmin 1 [100] | Embryonic inner ear and organotypic culture: Failed to rescue Ca2+-influx-triggered exocytosis | DB-OTO phase 1/2 clinical trial in pediatric patients |
| Dual AAV-Otof [103] | P10-RWM injection Total and sustained rescue ABR threshold shifts Amplitude wave I: 39% of the WT (P10injection), 50% of the WT (P17 injection) Ribbon number twice higher> non treated, but <WT | |||
| Single overloaded AAV-Otof [105] | P5-7 RWM injection: Expression of otoferlin in 30% of IHCs Partial restoration of hearing Poor preservation of wave I | |||
| DFNA25 | SLC17A8 | AAV1- Slc17a8 [36] | P1-P2 RW injection: 100% recovery ABR thresholds 40% sustained ABR recovery | |
| AAV8- Slc17a8 [106] | 5 w, 8 w, and 20 w canalostomic injection: 5 w injection: restore Vglut3 expression and hearing Partially restore the number of synapses 8 w injection: partial rescue of hearing 20 w injection: rescue less than 50% of ABR threshold | |||
| DFNB93 | CABP2 | AAV2/1 and PHP.eB-CABP2 [51] | P5-7 RW injection: Improve at least 20dB in all frequencies in 67% of the injected mice | |
| CMT | MPZ PMP22 | RNA-interference (RNAi) [110] AAV2/9 -Pmp22 shRNA [111] miR-318 [112] CRISPR/Cas9 [116] PMP22 antisense [115] | Intraneural injections: Normalize MPZ and PMP22 protein levels Improve myelination, function, locomotor activity, and electrophysiological parameters Subcutaneous injection: Reduce the mRNA levels of Pmp22, improve functional and morphological abnormalities of CMT1A | |
| NT-3 supplementation [119] | Subcutaneous injection: Improve axonal regeneration | |||
| AAV1-NT-3 cDNA [120] | Intramuscular injection: Improve motor function, histopathology, and electrophysiology of peripheral nerves | phase I/IIa clinical trial (NCT03520751) | ||
| DAO | OPA | AAV2-OPA1 [122]. | Intravitreal gene delivery Reduce retinal ganglion cell degeneration without rescuing an efficient visual acuity | |
| U1 splice factors [123] (bind to intron 10 at position +18 of OPA1) | In vitro: patient-derived and control fibroblasts Silence the effect of the mutation, increase the expression level of normal transcripts | |||
| CRISPR/Cas9–iPSCs (c.1334G>A: p.R445H) [124] | In vitro: Restore mitochondrial homeostasis, re-establish the mitochondrial network, basal respiration, and ATP production levels | |||
| LHON | Mt DNA TMEM126A | rAAV5-NDI1 [128] | Stereotaxic injections: infusion into the optical layer of the SC Rescue vision loss induced by complex I deficiency | |
| AAV2-NDI1 [131] | Intravitreal gene delivery: Mitochondrial internalization of AAVV Reduce RGC death and optic nerve atrophy Preserve retinal function (manganese, Mn2 þ)-enhanced magnetic resonance imaging (MEMRI) and optokinetic responses | Phase 1 clinical trial of scAAV2-P1ND4v2 of ND4-LHON (NCT02161380) | ||
| AAV2-ND4 [125,127,129,133] | Restore the activity of the respiratory chain and rescuing retinal ganglion cell degeneration | Phase 3 pivotal clinical study of rAAV2/2-ND4: REFLECT (NCT03293524) |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Starr, A.; Picton, T.W.; Sininger, Y.; Hood, L.J.; Berlin, C.I. Auditory Neuropathy. Brain 1996, 119, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Starr, A.; Michalewski, H.J.; Zeng, F.-G.; Fujikawa-Brooks, S.; Linthicum, F.; Kim, C.S.; Winnier, D.; Keats, B. Pathology and Physiology of Auditory Neuropathy with a Novel Mutation in the MPZ Gene (Tyr145->Ser). Brain 2003, 126, 1604–1619. [Google Scholar] [CrossRef] [PubMed]
- Amatuzzi, M.G.; Northrop, C.; Liberman, M.C.; Thornton, A.; Halpin, C.; Herrmann, B.; Pinto, L.E.; Saenz, A.; Carranza, A.; Eavey, R.D. Selective Inner Hair Cell Loss in Premature Infants and Cochlea Pathological Patterns from Neonatal Intensive Care Unit Autopsies. Arch. Otolaryngol.-Head Neck Surg. 2001, 127, 629–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blegvad, B.; Hvidegaard, T. Hereditary Dysfunction of the Brain Stem Auditory Pathways as the Major Cause of Speech Retardation. Scand. Audiol. 1983, 12, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.; Starr, A. Auditory Neuropathy—Neural and Synaptic Mechanisms. Nat. Rev. Neurol. 2016, 12, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Iliadou, V.V.; Ptok, M.; Grech, H.; Pedersen, E.R.; Brechmann, A.A.; Deggouj, N.N.; Kiese-Himmel, C.; Sliwinska-Kowalska, M.; Nickisch, A.; Demanez, L.; et al. A European Perspective on Auditory Processing Disorder-Current Knowledge and Future Research Focus. Front. Neurol. 2017, 8, 622. [Google Scholar] [CrossRef] [Green Version]
- Starr, A.; McPherson, D.; Patterson, J.; Don, M.; Luxford, W.; Shannon, R.; Sininger, Y.; Tonakawa, L.; Waring, M. Absence of Both Auditory Evoked Potentials and Auditory Percepts Dependent on Timing Cues. Brain 1991, 114, 1157–1180. [Google Scholar] [CrossRef] [Green Version]
- Starr, A.; Sininger, Y.; Nguyen, T.; Michalewski, H.J.; Oba, S.; Abdala, C. Cochlear Receptor (Microphonic and Summating Potentials, Otoacoustic Emissions) and Auditory Pathway (Auditory Brain Stem Potentials) Activity in Auditory Neuropathy. Ear Hear. 2001, 22, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Wynne, D.P.; Zeng, F.-G.; Bhatt, S.; Michalewski, H.J.; Dimitrijevic, A.; Starr, A. Loudness Adaptation Accompanying Ribbon Synapse and Auditory Nerve Disorders. Brain 2013, 136 Pt 5, 1626–1638. [Google Scholar] [CrossRef] [Green Version]
- Zeng, F.G.; Shannon, R.V. Loudness-Coding Mechanisms Inferred from Electric Stimulation of the Human Auditory System. Science 1994, 264, 564–566. [Google Scholar] [CrossRef]
- Zeng, F.-G. An Active Loudness Model Suggesting Tinnitus as Increased Central Noise and Hyperacusis as Increased Nonlinear Gain. Hear. Res. 2013, 295, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Siati, R.D.; Rosenzweig, F.; Gersdorff, G.; Gregoire, A.; Rombaux, P.; Deggouj, N. Auditory Neuropathy Spectrum Disorders: From Diagnosis to Treatment: Literature Review and Case Reports. JCM 2020, 9, 1074. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, S.; Wanna, G.; Hayes, C.; Sunderhaus, L.; Haynes, D.S.; Bennett, M.L.; Labadie, R.F.; Rivas, A. Cochlear Implantation versus Hearing Amplification in Patients with Auditory Neuropathy Spectrum Disorder. Otolaryngol.-Head Neck Surg. 2013, 148, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.; McCreery, R.; Spratford, M.; Roush, P. Children with Auditory Neuropathy Spectrum Disorder Fitted with Hearing Aids Applying the American Academy of Audiology Pediatric Amplification Guideline: Current Practice and Outcomes. J. Am. Acad. Audiol. 2016, 27, 204–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphriss, R.; Hall, A.; Maddocks, J.; Macleod, J.; Sawaya, K.; Midgley, E. Does Cochlear Implantation Improve Speech Recognition in Children with Auditory Neuropathy Spectrum Disorder? A Systematic Review. Int. J. Audiol. 2013, 52, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Ching, T.Y.C.; Day, J.; Dillon, H.; Gardner-Berry, K.; Hou, S.; Seeto, M.; Wong, A.; Zhang, V. Impact of the Presence of Auditory Neuropathy Spectrum Disorder (ANSD) on Outcomes of Children at Three Years of Age. Int. J. Audiol. 2013, 52 (Suppl. S2), S55–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarelli, R.; Rossi, R.; Scimemi, P.; Cama, E.; Valentino, M.L.; La Morgia, C.; Caporali, L.; Liguori, R.; Magnavita, V.; Monteleone, A.; et al. OPA1-Related Auditory Neuropathy: Site of Lesion and Outcome of Cochlear Implantation. Brain 2015, 138, 563–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roush, P.; Frymark, T.; Venediktov, R.; Wang, B. Audiologic Management of Auditory Neuropathy Spectrum Disorder in Children: A Systematic Review of the Literature. Am. J. Audiol. 2011, 20, 159–170. [Google Scholar] [CrossRef]
- Johnson, C.P.; Chapman, E.R. Otoferlin Is a Calcium Sensor That Directly Regulates SNARE-Mediated Membrane Fusion. J. Cell Biol. 2010, 191, 187–197. [Google Scholar] [CrossRef]
- Michalski, N.; Goutman, J.D.; Auclair, S.M.; Boutet de Monvel, J.; Tertrais, M.; Emptoz, A.; Parrin, A.; Nouaille, S.; Guillon, M.; Sachse, M.; et al. Otoferlin Acts as a Ca2+ Sensor for Vesicle Fusion and Vesicle Pool Replenishment at Auditory Hair Cell Ribbon Synapses. Elife 2017, 6, e31013. [Google Scholar] [CrossRef]
- Pangršič, T.; Reisinger, E.; Moser, T. Otoferlin: A Multi-C2 Domain Protein Essential for Hearing. Trends Neurosci. 2012, 35, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, S.; Grati, M.; Cohen-Salmon, M.; El-Amraoui, A.; Mustapha, M.; Salem, N.; El-Zir, E.; Loiselet, J.; Petit, C. A Mutation in OTOF, Encoding Otoferlin, a FER-1-like Protein, Causes DFNB9, a Nonsyndromic Form of Deafness. Nat. Genet. 1999, 21, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Vona, B.; Doll, J.; Hofrichter, M.A.H.; Haaf, T.; Varshney, G.K. Small Fish, Big Prospects: Using Zebrafish to Unravel the Mechanisms of Hereditary Hearing Loss. Hear. Res. 2020, 397, 107906. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.; del Castillo, I.; Cama, E.; Scimemi, P.; Starr, A. Audibility, Speech Perception and Processing of Temporal Cues in Ribbon Synaptic Disorders Due to OTOF Mutations. Hear. Res. 2015, 330, 200–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.Y.; Ahmed, Z.M.; Riazuddin, S.; Bhinder, M.A.; Shahzad, M.; Husnain, T.; Riazuddin, S.; Griffith, A.J.; Friedman, T.B. Identities and Frequencies of Mutations of the Otoferlin Gene (OTOF) Causing DFNB9 Deafness in Pakistan. Clin. Genet. 2009, 75, 237–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Ballesteros, M.; Reynoso, R.; Olarte, M.; Villamar, M.; Morera, C.; Santarelli, R.; Arslan, E.; Medá, C.; Curet, C.; Völter, C.; et al. A Multicenter Study on the Prevalence and Spectrum of Mutations in the Otoferlin Gene (OTOF) in Subjects with Nonsyndromic Hearing Impairment and Auditory Neuropathy. Hum. Mutat. 2008, 29, 823–831. [Google Scholar] [CrossRef]
- Varga, R.; Avenarius, M.R.; Kelley, P.M.; Keats, B.J.; Berlin, C.I.; Hood, L.J.; Morlet, T.G.; Brashears, S.M.; Starr, A.; Cohn, E.S.; et al. OTOF Mutations Revealed by Genetic Analysis of Hearing Loss Families Including a Potential Temperature Sensitive Auditory Neuropathy Allele. J. Med. Genet. 2006, 43, 576–581. [Google Scholar] [CrossRef] [Green Version]
- Santarelli, R.; Scimemi, P.; La Morgia, C.; Cama, E.; Del Castillo, I.; Carelli, V. Electrocochleography in Auditory Neuropathy Related to Mutations in the OTOF or OPA1 Gene. Audiol. Res. 2021, 11, 639–652. [Google Scholar] [CrossRef]
- Pangrsic, T.; Lasarow, L.; Reuter, K.; Takago, H.; Schwander, M.; Riedel, D.; Frank, T.; Tarantino, L.M.; Bailey, J.S.; Strenzke, N.; et al. Hearing Requires Otoferlin-Dependent Efficient Replenishment of Synaptic Vesicles in Hair Cells. Nat. Neurosci. 2010, 13, 869–876. [Google Scholar] [CrossRef]
- Roux, I.; Safieddine, S.; Nouvian, R.; Grati, M.; Simmler, M.-C.; Bahloul, A.; Perfettini, I.; Le Gall, M.; Rostaing, P.; Hamard, G.; et al. Otoferlin, Defective in a Human Deafness Form, Is Essential for Exocytosis at the Auditory Ribbon Synapse. Cell 2006, 127, 277–289. [Google Scholar] [CrossRef]
- El Mestikawy, S.; Wallén-Mackenzie, A.; Fortin, G.M.; Descarries, L.; Trudeau, L.-E. From Glutamate Co-Release to Vesicular Synergy: Vesicular Glutamate Transporters. Nat. Rev. Neurosci. 2011, 12, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, X.; Cao, H.; Zhao, H.; Geller, A.I. The Vesicular Glutamate Transporter-1 Upstream Promoter and First Intron Each Support Glutamatergic-Specific Expression in Rat Postrhinal Cortex. Brain Res. 2011, 1377, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruel, J.; Emery, S.; Nouvian, R.; Bersot, T.; Amilhon, B.; Van Rybroek, J.M.; Rebillard, G.; Lenoir, M.; Eybalin, M.; Delprat, B.; et al. Impairment of SLC17A8 Encoding Vesicular Glutamate Transporter-3, VGLUT3, Underlies Nonsyndromic Deafness DFNA25 and Inner Hair Cell Dysfunction in Null Mice. Am. J. Hum. Genet. 2008, 83, 278–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seal, R.P.; Akil, O.; Yi, E.; Weber, C.M.; Grant, L.; Yoo, J.; Clause, A.; Kandler, K.; Noebels, J.L.; Glowatzki, E.; et al. Sensorineural Deafness and Seizures in Mice Lacking Vesicular Glutamate Transporter 3. Neuron 2008, 57, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Weisz, C.J.C.; Williams, S.-P.G.; Eckard, C.S.; Divito, C.B.; Ferreira, D.W.; Fantetti, K.N.; Dettwyler, S.A.; Cai, H.-M.; Rubio, M.E.; Kandler, K.; et al. Outer Hair Cell Glutamate Signaling through Type II Spiral Ganglion Afferents Activates Neurons in the Cochlear Nucleus in Response to Nondamaging Sounds. J. Neurosci. 2021, 41, 2930–2943. [Google Scholar] [CrossRef]
- Akil, O.; Seal, R.P.; Burke, K.; Wang, C.; Alemi, A.; During, M.; Edwards, R.H.; Lustig, L.R. Restoration of Hearing in the VGLUT3 Knockout Mouse Using Virally Mediated Gene Therapy. Neuron 2012, 75, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.X.; Payne, S.; Yang-Hood, A.; Li, S.-Z.; Davis, B.; Carlquist, J.; V.-Ghaffari, B.; Gantz, J.A.; Kallogjeri, D.; Fitzpatrick, J.A.J.; et al. Vesicular Glutamatergic Transmission in Noise-Induced Loss and Repair of Cochlear Ribbon Synapses. J. Neurosci. 2019, 39, 4434–4447. [Google Scholar] [CrossRef] [Green Version]
- Petek, E.; Windpassinger, C.; Mach, M.; Rauter, L.; Scherer, S.W.; Wagner, K.; Kroisel, P.M. Molecular Characterization of a 12q22-Q24 Deletion Associated with Congenital Deafness: Confirmation and Refinement of the DFNA25 Locus. Am. J. Med. Genet. Part A 2003, 117, 122–126. [Google Scholar] [CrossRef]
- Ryu, N.; Sagong, B.; Park, H.-J.; Kim, M.-A.; Lee, K.-Y.; Choi, J.Y.; Kim, U.-K. Screening of the SLC17A8 Gene as a Causative Factor for Autosomal Dominant Non-Syndromic Hearing Loss in Koreans. BMC Med. Genet. 2016, 17, 6. [Google Scholar] [CrossRef] [Green Version]
- Joshi, Y.; Petit, C.P.; Miot, S.; Guillet, M.; Sendin, G.; Bourien, J.; Wang, J.; Pujol, R.; El Mestikawy, S.; Puel, J.-L.; et al. VGLUT3-p.A211V Variant Fuses Stereocilia Bundles and Elongates Synaptic Ribbons. J. Physiol. 2021, 599, 5397–5416. [Google Scholar] [CrossRef]
- Brandt, A.; Striessnig, J.; Moser, T. CaV1.3 Channels Are Essential for Development and Presynaptic Activity of Cochlear Inner Hair Cells. J. Neurosci. 2003, 23, 10832–10840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, S.M.; Koschak, A.; Lieb, A.; Gebhart, M.; Dafinger, C.; Nürnberg, G.; Ali, A.; Ahmad, I.; Sinnegger-Brauns, M.J.; Brandt, N.; et al. Loss of Ca(v)1.3 (CACNA1D) Function in a Human Channelopathy with Bradycardia and Congenital Deafness. Nat. Neurosci. 2011, 14, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Platzer, J.; Engel, J.; Schrott-Fischer, A.; Stephan, K.; Bova, S.; Chen, H.; Zheng, H.; Striessnig, J. Congenital Deafness and Sinoatrial Node Dysfunction in Mice Lacking Class D L-Type Ca2+ Channels. Cell 2000, 102, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, F.; Zhang, R.; Chen, J.; Zhao, F.; Sun, Y.; Du, Z.; Bing, D.; Li, P.; Shao, S.; Zhu, H.; et al. Down-Regulation of Cav1.3 in Auditory Pathway Promotes Age-Related Hearing Loss by Enhancing Calcium-Mediated Oxidative Stress in Male Mice. Aging 2019, 11, 6490–6502. [Google Scholar] [CrossRef] [PubMed]
- Eckrich, S.; Hecker, D.; Sorg, K.; Blum, K.; Fischer, K.; Münkner, S.; Wenzel, G.; Schick, B.; Engel, J. Cochlea-Specific Deletion of Cav1.3 Calcium Channels Arrests Inner Hair Cell Differentiation and Unravels Pitfalls of Conditional Mouse Models. Front. Cell. Neurosci. 2019, 13, 225. [Google Scholar] [CrossRef]
- Haeseleer, F.; Imanishi, Y.; Sokal, I.; Filipek, S.; Palczewski, K. Calcium-Binding Proteins: Intracellular Sensors from the Calmodulin Superfamily. Biochem. Biophys. Res. Commun. 2002, 290, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Christel, C.; Lee, A. Ca2+-Dependent Modulation of Voltage-Gated Ca2+ Channels. Biochim. Biophys. Acta 2012, 1820, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Tabatabaiefar, M.A.; Alasti, F.; Shariati, L.; Farrokhi, E.; Fransen, E.; Nooridaloii, M.R.; Chaleshtori, M.H.; Van Camp, G. DFNB93, a Novel Locus for Autosomal Recessive Moderate-to-Severe Hearing Impairment. Clin. Genet. 2011, 79, 594–598. [Google Scholar] [CrossRef]
- Schrauwen, I.; Helfmann, S.; Inagaki, A.; Predoehl, F.; Tabatabaiefar, M.A.; Picher, M.M.; Sommen, M.; Zazo Seco, C.; Oostrik, J.; Kremer, H.; et al. A Mutation in CABP2, Expressed in Cochlear Hair Cells, Causes Autosomal-Recessive Hearing Impairment. Am. J. Hum. Genet. 2012, 91, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Bademci, G.; Foster, J.; Mahdieh, N.; Bonyadi, M.; Duman, D.; Cengiz, F.B.; Menendez, I.; Diaz-Horta, O.; Shirkavand, A.; Zeinali, S.; et al. Comprehensive Analysis via Exome Sequencing Uncovers Genetic Etiology in Autosomal Recessive Nonsyndromic Deafness in a Large Multiethnic Cohort. Genet. Med. 2016, 18, 364–371. [Google Scholar] [CrossRef]
- Oestreicher, D.; Picher, M.M.; Rankovic, V.; Moser, T.; Pangrsic, T. Cabp2-Gene Therapy Restores Inner Hair Cell Calcium Currents and Improves Hearing in a DFNB93 Mouse Model. Front. Mol. Neurosci. 2021, 14, 689415. [Google Scholar] [CrossRef] [PubMed]
- Picher, M.M.; Gehrt, A.; Meese, S.; Ivanovic, A.; Predoehl, F.; Jung, S.; Schrauwen, I.; Dragonetti, A.G.; Colombo, R.; Van Camp, G.; et al. Ca2+-Binding Protein 2 Inhibits Ca2+-Channel Inactivation in Mouse Inner Hair Cells. Proc. Natl. Acad. Sci. USA 2017, 114, E1717–E1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Hu, N.; Pangršič, T.; Green, S.; Hansen, M.; Lee, A. Functions of CaBP1 and CaBP2 in the Peripheral Auditory System. Hear. Res. 2018, 364, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Schoen, C.J.; Burmeister, M.; Lesperance, M.M. Diaphanous Homolog 3 (Diap3) Overexpression Causes Progressive Hearing Loss and Inner Hair Cell Defects in a Transgenic Mouse Model of Human Deafness. PLoS ONE 2013, 8, e56520. [Google Scholar] [CrossRef] [Green Version]
- Rance, G.; Starr, A. Pathophysiological Mechanisms and Functional Hearing Consequences of Auditory Neuropathy. Brain 2015, 138, 3141–3158. [Google Scholar] [CrossRef] [PubMed]
- Surel, C.; Guillet, M.; Lenoir, M.; Bourien, J.; Sendin, G.; Joly, W.; Delprat, B.; Lesperance, M.M.; Puel, J.-L.; Nouvian, R. Remodeling of the Inner Hair Cell Microtubule Meshwork in a Mouse Model of Auditory Neuropathy AUNA1. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Manchaiah, V.K.C.; Zhao, F.; Danesh, A.A.; Duprey, R. The Genetic Basis of Auditory Neuropathy Spectrum Disorder (ANSD). Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 151–158. [Google Scholar] [CrossRef]
- Rendtorff, N.D.; Lodahl, M.; Boulahbel, H.; Johansen, I.R.; Pandya, A.; Welch, K.O.; Norris, V.W.; Arnos, K.S.; Bitner-Glindzicz, M.; Emery, S.B.; et al. Identification of p.A684V Missense Mutation in the WFS1 Gene as a Frequent Cause of Autosomal Dominant Optic Atrophy and Hearing Impairment. Am. J. Med. Genet. Part A 2011, 155, 1298–1313. [Google Scholar] [CrossRef] [Green Version]
- Morlet, T.; Nagao, K.; Bean, S.C.; Mora, S.E.; Hopkins, S.E.; Hobson, G.M. Auditory Function in Pelizaeus-Merzbacher Disease. J. Neurol. 2018, 265, 1580–1589. [Google Scholar] [CrossRef]
- Han, K.-H.; Oh, D.-Y.; Lee, S.; Lee, C.; Han, J.H.; Kim, M.Y.; Park, H.-R.; Park, M.K.; Kim, N.K.D.; Lee, J.; et al. ATP1A3 Mutations Can Cause Progressive Auditory Neuropathy: A New Gene of Auditory Synaptopathy. Sci. Rep. 2017, 7, 16504. [Google Scholar] [CrossRef]
- Lupski, J.R.; de Oca-Luna, R.M.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA Duplication Associated with Charcot-Marie-Tooth Disease Type 1A. Cell 1991, 66, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Rance, G.; Ryan, M.M.; Bayliss, K.; Gill, K.; O’Sullivan, C.; Whitechurch, M. Auditory Function in Children with Charcot-Marie-Tooth Disease. Brain 2012, 135, 1412–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raglan, E.; Prasher, D.K.; Trinder, E.; Rudge, P. Auditory Function in Hereditary Motor and Sensory Neuropathy (Charcot-Marie-Tooth Disease). Acta Oto-Laryngol. 1987, 103, 50–55. [Google Scholar] [CrossRef]
- Kovach, M.J.; Campbell, K.C.M.; Herman, K.; Waggoner, B.; Gelber, D.; Hughes, L.F.; Kimonis, V.E. Anticipation in a Unique Family with Charcot-Marie-Tooth Syndrome and Deafness: Delineation of the Clinical Features and Review of the Literature. Am. J. Med. Genet. 2002, 108, 295–303. [Google Scholar] [CrossRef]
- Choi, J.E.; Seok, J.M.; Ahn, J.; Ji, Y.S.; Lee, K.M.; Hong, S.H.; Choi, B.-O.; Moon, I.J. Hidden Hearing Loss in Patients with Charcot-Marie-Tooth Disease Type 1A. Sci. Rep. 2018, 8, 10335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, P.B.; Gaster, R.N.; Smith, V.C.; Tripathi, R.C. A Clinicopathologic Study of Autosomal Dominant Optic Atrophy. Am. J. Ophthalmol. 1979, 88, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, Encoding a Dynamin-Related GTPase, Is Mutated in Autosomal Dominant Optic Atrophy Linked to Chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear Gene OPA1, Encoding a Mitochondrial Dynamin-Related Protein, Is Mutated in Dominant Optic Atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Lenaers, G.; Neutzner, A.; Le Dantec, Y.; Jüschke, C.; Xiao, T.; Decembrini, S.; Swirski, S.; Kieninger, S.; Agca, C.; Kim, U.S.; et al. Dominant Optic Atrophy: Culprit Mitochondria in the Optic Nerve. Prog. Retin. Eye Res. 2021, 83, 100935. [Google Scholar] [CrossRef]
- Amati-Bonneau, P.; Guichet, A.; Olichon, A.; Chevrollier, A.; Viala, F.; Miot, S.; Ayuso, C.; Odent, S.; Arrouet, C.; Verny, C.; et al. OPA1 R445H Mutation in Optic Atrophy Associated with Sensorineural Deafness. Ann. Neurol. 2005, 58, 958–963. [Google Scholar] [CrossRef]
- Lodi, R.; Tonon, C.; Valentino, M.L.; Iotti, S.; Clementi, V.; Malucelli, E.; Barboni, P.; Longanesi, L.; Schimpf, S.; Wissinger, B.; et al. Deficit of in Vivo Mitochondrial ATP Production in OPA1-Related Dominant Optic Atrophy. Ann. Neurol. 2004, 56, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 Perturbates the Mitochondrial Inner Membrane Structure and Integrity, Leading to Cytochrome c Release and Apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 Mutations Induce Mitochondrial DNA Instability and Optic Atrophy “plus” Phenotypes. Brain 2008, 131, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Belenguer, P.; Pellegrini, L. The Dynamin GTPase OPA1: More than Mitochondria? Biochim. Et Biophys. Acta 2013, 1833, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Elachouri, G.; Vidoni, S.; Zanna, C.; Pattyn, A.; Boukhaddaoui, H.; Gaget, K.; Yu-Wai-Man, P.; Gasparre, G.; Sarzi, E.; Delettre, C.; et al. OPA1 Links Human Mitochondrial Genome Maintenance to MtDNA Replication and Distribution. Genome Res. 2011, 21, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Mitochondrial Dysfunction as a Cause of Optic Neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89. [Google Scholar] [CrossRef]
- Baker, M.R.; Fisher, K.M.; Whittaker, R.G.; Griffiths, P.G.; Yu-Wai-Man, P.; Chinnery, P.F. Subclinical Multisystem Neurologic Disease in “Pure” OPA1 Autosomal Dominant Optic Atrophy. Neurology 2011, 77, 1309–1312. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-System Neurological Disease Is Common in Patients with OPA1 Mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef]
- Leruez, S.; Milea, D.; Defoort-Dhellemmes, S.; Colin, E.; Crochet, M.; Procaccio, V.; Ferré, M.; Lamblin, J.; Drouin, V.; Vincent-Delorme, C.; et al. Sensorineural Hearing Loss in OPA1-Linked Disorders. Brain 2013, 136, e236. [Google Scholar] [CrossRef]
- Liskova, P.; Ulmanova, O.; Tesina, P.; Melsova, H.; Diblik, P.; Hansikova, H.; Tesarova, M.; Votruba, M. Novel OPA1 Missense Mutation in a Family with Optic Atrophy and Severe Widespread Neurological Disorder. Acta Ophthalmol. 2013, 91, e225–e231. [Google Scholar] [CrossRef] [PubMed]
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant Optic Atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Santarelli, R.; Starr, A. Mutation of OPA1 Gene Causes Deafness by Affecting Function of Auditory Nerve Terminals. Brain Res. 2009, 1300, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Santarelli, R. Information from Cochlear Potentials and Genetic Mutations Helps Localize the Lesion Site in Auditory Neuropathy. Genome Med. 2010, 2, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.H.; Blackwood, W.; Wilson, J. Further Clinical and Pathological Observations on Leber’s Optic Atrophy. Brain 1966, 89, 15–26. [Google Scholar] [CrossRef]
- Ceranić, B.; Luxon, L.M. Progressive Auditory Neuropathy in Patients with Leber’s Hereditary Optic Neuropathy. J. Neurol. Neurosurg. Psychiatry 2004, 75, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Meyer, E.; Michaelides, M.; Tee, L.J.; Robson, A.G.; Rahman, F.; Pasha, S.; Luxon, L.M.; Moore, A.T.; Maher, E.R. Nonsense Mutation in TMEM126A Causing Autosomal Recessive Optic Atrophy and Auditory Neuropathy. Mol. Vis. 2010, 16, 650–664. [Google Scholar]
- Dürr, A.; Cossee, M.; Agid, Y.; Campuzano, V.; Mignard, C.; Penet, C.; Mandel, J.L.; Brice, A.; Koenig, M. Clinical and Genetic Abnormalities in Patients with Friedreich’s Ataxia. N. Engl. J. Med. 1996, 335, 1169–1175. [Google Scholar] [CrossRef]
- Harding, A.E. Friedreich’s Ataxia: A Clinical and Genetic Study of 90 Families with an Analysis of Early Diagnostic Criteria and Intrafamilial Clustering of Clinical Features. Brain 1981, 104, 589–620. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, J.M.; Balcer, L.J.; Wilson, R.B. Friedreich Ataxia: Effects of Genetic Understanding on Clinical Evaluation and Therapy. Arch. Neurol. 2002, 59, 743–747. [Google Scholar] [CrossRef] [Green Version]
- Rance, G.; Barker, E.J. Speech Perception in Children with Auditory Neuropathy/Dyssynchrony Managed with Either Hearing AIDS or Cochlear Implants. Otol. Neurotol. 2008, 29, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Rance, G.; Corben, L.; Barker, E.; Carew, P.; Chisari, D.; Rogers, M.; Dowell, R.; Jamaluddin, S.; Bryson, R.; Delatycki, M.B. Auditory Perception in Individuals with Friedreich’s Ataxia. Audiol. Neurotol. 2010, 15, 229–240. [Google Scholar] [CrossRef]
- Jabbari, B.; Schwartz, D.M.; MacNeil, D.M.; Coker, S.B. Early Abnormalities of Brainstem Auditory Evoked Potentials in Friedreich’s Ataxia: Evidence of Primary Brainstem Dysfunction. Neurology 1983, 33, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.; Cama, E.; Pegoraro, E.; Scimemi, P. Abnormal Cochlear Potentials in Friedreich’s Ataxia Point to Disordered Synchrony of Auditory Nerve Fiber Activity. Neurodegener. Dis. 2015, 15, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Koohi, N.; Thomas-Black, G.; Giunti, P.; Bamiou, D.-E. Auditory Phenotypic Variability in Friedreich’s Ataxia Patients. Cerebellum 2021, 20, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Bahmad, F., Jr.; Merchant, S.N.; Nadol, J.B., Jr.; Tranebjærg, L. Otopathology in Mohr-Tranebjærg Syndrome. Laryngoscope 2007, 117, 1202–1208. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, R.C. The Basic Science of Gene Therapy. Science 1993, 260, 926–932. [Google Scholar] [CrossRef]
- Sacheli, R.; Delacroix, L.; Vandenackerveken, P.; Nguyen, L.; Malgrange, B. Gene Transfer in Inner Ear Cells: A Challenging Race. Gene Ther. 2013, 20, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Puel, J.-L. Toward Cochlear Therapies. Physiol. Rev. 2018, 98, 2477–2522. [Google Scholar] [CrossRef]
- Reisinger, E.; Bresee, C.; Neef, J.; Nair, R.; Reuter, K.; Bulankina, A.; Nouvian, R.; Koch, M.; Bückers, J.; Kastrup, L.; et al. Probing the Functional Equivalence of Otoferlin and Synaptotagmin 1 in Exocytosis. J. Neurosci. 2011, 31, 4886–4895. [Google Scholar] [CrossRef] [Green Version]
- Beurg, M.; Michalski, N.; Safieddine, S.; Bouleau, Y.; Schneggenburger, R.; Chapman, E.R.; Petit, C.; Dulon, D. Control of Exocytosis by Synaptotagmins and Otoferlin in Auditory Hair Cells. J. Neurosci. 2010, 30, 13281–13290. [Google Scholar] [CrossRef] [PubMed]
- Safieddine, S.; Wenthold, R.J. SNARE Complex at the Ribbon Synapses of Cochlear Hair Cells: Analysis of Synaptic Vesicle- and Synaptic Membrane-Associated Proteins. Eur. J. Neurosci. 1999, 11, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akil, O.; Dyka, F.; Calvet, C.; Emptoz, A.; Lahlou, G.; Nouaille, S.; Boutet de Monvel, J.; Hardelin, J.-P.; Hauswirth, W.W.; Avan, P.; et al. Dual AAV-Mediated Gene Therapy Restores Hearing in a DFNB9 Mouse Model. Proc. Natl. Acad. Sci. USA 2019, 116, 4496–4501. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Yue, Y.; Duan, D. Efficient Transgene Reconstitution with Hybrid Dual AAV Vectors Carrying the Minimized Bridging Sequences. Hum. Gene Ther. 2011, 22, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, V.; Vogl, C.; Dörje, N.M.; Bahader, I.; Duque-Afonso, C.J.; Thirumalai, A.; Weber, T.; Kusch, K.; Strenzke, N.; Moser, T. Overloaded Adeno-Associated Virus as a Novel Gene Therapeutic Tool for Otoferlin-Related Deafness. Front. Mol. Neurosci. 2020, 13, 600051. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, H.; Liu, H.; Cai, R.; Wu, H. Gene Therapy Restores Auditory Functions in an Adult Vglut3 Knockout Mouse Model. Hum. Gene Ther. 2022, 33, 729–739. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Bolhuis, P.A.; Zorn, I.; Hoogendijk, J.E.; van den Bosch, N.; Hensels, G.W.; Stanton, V.P.; Housman, D.E.; Fischbeck, K.H.; Ross, D.A. The Peripheral Myelin Gene PMP-22/GAS-3 Is Duplicated in Charcot-Marie-Tooth Disease Type 1A. Nat. Genet. 1992, 1, 166–170. [Google Scholar] [CrossRef]
- Timmerman, V.; Nelis, E.; Van Hul, W.; Nieuwenhuijsen, B.W.; Chen, K.L.; Wang, S.; Ben Othman, K.; Cullen, B.; Leach, R.J.; Hanemann, C.O. The Peripheral Myelin Protein Gene PMP-22 Is Contained within the Charcot-Marie-Tooth Disease Type 1A Duplication. Nat. Genet. 1992, 1, 171–175. [Google Scholar] [CrossRef]
- Matsunami, N.; Smith, B.; Ballard, L.; Lensch, M.W.; Robertson, M.; Albertsen, H.; Hanemann, C.O.; Müller, H.W.; Bird, T.D.; White, R. Peripheral Myelin Protein-22 Gene Maps in the Duplication in Chromosome 17p11.2 Associated with Charcot-Marie-Tooth 1A. Nat. Genet. 1992, 1, 176–179. [Google Scholar] [CrossRef]
- Lee, J.-S.; Chang, E.H.; Koo, O.J.; Jwa, D.H.; Mo, W.M.; Kwak, G.; Moon, H.W.; Park, H.T.; Hong, Y.B.; Choi, B.-O. Pmp22 Mutant Allele-Specific SiRNA Alleviates Demyelinating Neuropathic Phenotype in Vivo. Neurobiol. Dis. 2017, 100, 99–107. [Google Scholar] [CrossRef]
- Gautier, B.; Hajjar, H.; Soares, S.; Berthelot, J.; Deck, M.; Abbou, S.; Campbell, G.; Ceprian, M.; Gonzalez, S.; Fovet, C.-M.; et al. AAV2/9-Mediated Silencing of PMP22 Prevents the Development of Pathological Features in a Rat Model of Charcot-Marie-Tooth Disease 1 A. Nat. Commun. 2021, 12, 2356. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Kwak, G.; Kim, H.J.; Park, H.-T.; Choi, B.-O.; Hong, Y.B. MiR-381 Attenuates Peripheral Neuropathic Phenotype Caused by Overexpression of PMP22. Exp. Neurobiol. 2019, 28, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Boutary, S.; Caillaud, M.; El Madani, M.; Vallat, J.-M.; Loisel-Duwattez, J.; Rouyer, A.; Richard, L.; Gracia, C.; Urbinati, G.; Desmaële, D.; et al. Squalenoyl SiRNA PMP22 Nanoparticles Are Effective in Treating Mouse Models of Charcot-Marie-Tooth Disease Type 1 A. Commun. Biol. 2021, 4, 317. [Google Scholar] [CrossRef]
- Serfecz, J.; Bazick, H.; Al Salihi, M.O.; Turner, P.; Fields, C.; Cruz, P.; Renne, R.; Notterpek, L. Downregulation of the Human Peripheral Myelin Protein 22 Gene by MiR-29a in Cellular Models of Charcot-Marie-Tooth Disease. Gene Ther. 2019, 26, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.T.; Damle, S.; Ikeda-Lee, K.; Kuntz, S.; Li, J.; Mohan, A.; Kim, A.; Hung, G.; Scheideler, M.A.; Scherer, S.S.; et al. PMP22 Antisense Oligonucleotides Reverse Charcot-Marie-Tooth Disease Type 1A Features in Rodent Models. J. Clin. Investig. 2018, 128, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Lee, J.Y.; Song, D.W.; Bae, H.S.; Doo, H.M.; Yu, H.S.; Lee, K.J.; Kim, H.K.; Hwang, H.; Kwak, G.; et al. Targeted PMP22 TATA-Box Editing by CRISPR/Cas9 Reduces Demyelinating Neuropathy of Charcot-Marie-Tooth Disease Type 1A in Mice. Nucleic Acids Res. 2020, 48, 130–140. [Google Scholar] [CrossRef]
- Berger, A.; Maire, S.; Gaillard, M.-C.; Sahel, J.-A.; Hantraye, P.; Bemelmans, A.-P. MRNA Trans-Splicing in Gene Therapy for Genetic Diseases. Wiley Interdiscip. Rev. RNA 2016, 7, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Sahenk, Z.; Ozes, B. Gene Therapy to Promote Regeneration in Charcot-Marie-Tooth Disease. Brain Res. 2020, 1727, 146533. [Google Scholar] [CrossRef]
- Sahenk, Z.; Nagaraja, H.N.; McCracken, B.S.; King, W.M.; Freimer, M.L.; Cedarbaum, J.M.; Mendell, J.R. NT-3 Promotes Nerve Regeneration and Sensory Improvement in CMT1A Mouse Models and in Patients. Neurology 2005, 65, 681–689. [Google Scholar] [CrossRef]
- Sahenk, Z.; Galloway, G.; Clark, K.R.; Malik, V.; Rodino-Klapac, L.R.; Kaspar, B.K.; Chen, L.; Braganza, C.; Montgomery, C.; Mendell, J.R. AAV1.NT-3 Gene Therapy for Charcot-Marie-Tooth Neuropathy. Mol. Ther. 2014, 22, 511–521. [Google Scholar] [CrossRef]
- Marchbank, N.J.; Craig, J.E.; Leek, J.P.; Toohey, M.; Churchill, A.J.; Markham, A.F.; Mackey, D.A.; Toomes, C.; Inglehearn, C.F. Deletion of the OPA1 Gene in a Dominant Optic Atrophy Family: Evidence That Haploinsufficiency Is the Cause of Disease. J. Med. Genet. 2002, 39, e47. [Google Scholar] [CrossRef] [PubMed]
- Sarzi, E.; Seveno, M.; Piro-Mégy, C.; Elzière, L.; Quilès, M.; Péquignot, M.; Müller, A.; Hamel, C.P.; Lenaers, G.; Delettre, C. OPA1 Gene Therapy Prevents Retinal Ganglion Cell Loss in a Dominant Optic Atrophy Mouse Model. Sci. Rep. 2018, 8, 2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jüschke, C.; Klopstock, T.; Catarino, C.B.; Owczarek-Lipska, M.; Wissinger, B.; Neidhardt, J. Autosomal Dominant Optic Atrophy: A Novel Treatment for OPA1 Splice Defects Using U1 SnRNA Adaption. Mol. Ther. Nucleic Acids 2021, 26, 1186–1197. [Google Scholar] [CrossRef]
- Sladen, P.E.; Perdigão, P.R.L.; Salsbury, G.; Novoselova, T.; van der Spuy, J.; Chapple, J.P.; Yu-Wai-Man, P.; Cheetham, M.E. CRISPR-Cas9 Correction of OPA1 c.1334G>A: P.R445H Restores Mitochondrial Homeostasis in Dominant Optic Atrophy Patient-Derived IPSCs. Mol. Ther. Nucleic Acids 2021, 26, 432–443. [Google Scholar] [CrossRef]
- Sahel, J.-A.; Newman, N.J.; Yu-Wai-Man, P.; Vignal-Clermont, C.; Carelli, V.; Biousse, V.; Moster, M.L.; Sergott, R.; Klopstock, T.; Sadun, A.A.; et al. Gene Therapies for the Treatment of Leber Hereditary Optic Neuropathy. Int. Ophthalmol. Clin. 2021, 61, 195–208. [Google Scholar] [CrossRef]
- Hage, R.; Vignal-Clermont, C. Leber Hereditary Optic Neuropathy: Review of Treatment and Management. Front. Neurol. 2021, 12, 651639. [Google Scholar] [CrossRef] [PubMed]
- Ellouze, S.; Augustin, S.; Bouaita, A.; Bonnet, C.; Simonutti, M.; Forster, V.; Picaud, S.; Sahel, J.-A.; Corral-Debrinski, M. Optimized Allotopic Expression of the Human Mitochondrial ND4 Prevents Blindness in a Rat Model of Mitochondrial Dysfunction. Am. J. Hum. Genet. 2008, 83, 373–387. [Google Scholar] [CrossRef] [Green Version]
- Marella, M.; Seo, B.B.; Thomas, B.B.; Matsuno-Yagi, A.; Yagi, T. Successful Amelioration of Mitochondrial Optic Neuropathy Using the Yeast NDI1 Gene in a Rat Animal Model. PLoS ONE 2010, 5, e11472. [Google Scholar] [CrossRef]
- Shi, H.; Gao, J.; Pei, H.; Liu, R.; Hu, W.; Wan, X.; Li, T.; Li, B. Adeno-Associated Virus-Mediated Gene Delivery of the Human ND4 Complex I Subunit in Rabbit Eyes. Clin. Exp. Ophthalmol. 2012, 40, 888–894. [Google Scholar] [CrossRef]
- Yu, H.; Koilkonda, R.D.; Chou, T.-H.; Porciatti, V.; Ozdemir, S.S.; Chiodo, V.; Boye, S.L.; Boye, S.E.; Hauswirth, W.W.; Lewin, A.S.; et al. Gene Delivery to Mitochondria by Targeting Modified Adenoassociated Virus Suppresses Leber’s Hereditary Optic Neuropathy in a Mouse Model. Proc. Natl. Acad. Sci. USA 2012, 109, E1238–E1247. [Google Scholar] [CrossRef] [Green Version]
- Chadderton, N.; Palfi, A.; Millington-Ward, S.; Gobbo, O.; Overlack, N.; Carrigan, M.; O’Reilly, M.; Campbell, M.; Ehrhardt, C.; Wolfrum, U.; et al. Intravitreal Delivery of AAV-NDI1 Provides Functional Benefit in a Murine Model of Leber Hereditary Optic Neuropathy. Eur. J. Hum. Genet. 2013, 21, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Koilkonda, R.D.; Yu, H.; Chou, T.-H.; Feuer, W.J.; Ruggeri, M.; Porciatti, V.; Tse, D.; Hauswirth, W.W.; Chiodo, V.; Boye, S.L.; et al. Safety and Effects of the Vector for the Leber Hereditary Optic Neuropathy Gene Therapy Clinical Trial. JAMA Ophthalmol. 2014, 132, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Koilkonda, R.; Yu, H.; Talla, V.; Porciatti, V.; Feuer, W.J.; Hauswirth, W.W.; Chiodo, V.; Erger, K.E.; Boye, S.L.; Lewin, A.S.; et al. LHON Gene Therapy Vector Prevents Visual Loss and Optic Neuropathy Induced by G11778A Mutant Mitochondrial DNA: Biodistribution and Toxicology Profile. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7739–7753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.-H.; Pan, B.; Hu, Y.-J.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of Autosomal Dominant Hearing Loss by in Vivo Delivery of Genome Editing Agents. Nature 2018, 553, 217–221. [Google Scholar] [CrossRef]
- Hauswirth, W.W.; Aleman, T.S.; Kaushal, S.; Cideciyan, A.V.; Schwartz, S.B.; Wang, L.; Conlon, T.J.; Boye, S.L.; Flotte, T.R.; Byrne, B.J.; et al. Treatment of Leber Congenital Amaurosis Due to RPE65 Mutations by Ocular Subretinal Injection of Adeno-Associated Virus Gene Vector: Short-Term Results of a Phase I Trial. Hum. Gene Ther. 2008, 19, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Bainbridge, J.W.B.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of Gene Therapy on Visual Function in Leber’s Congenital Amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Aleman, T.S.; Boye, S.L.; Schwartz, S.B.; Kaushal, S.; Roman, A.J.; Pang, J.-J.; Sumaroka, A.; Windsor, E.A.M.; Wilson, J.M.; et al. Human Gene Therapy for RPE65 Isomerase Deficiency Activates the Retinoid Cycle of Vision but with Slow Rod Kinetics. Proc. Natl. Acad. Sci. USA 2008, 105, 15112–15117. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-Associated Virus Vector as a Platform for Gene Therapy Delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- HHTM. Decibel Therapeutics Announces Submission of Clinical Trial Applications for Lead Gene Therapy Candidate DB-OTO–HHTM. Hearing News Watch. Available online: https://hearinghealthmatters.org/hearingnewswatch/2022/decibel-therapeutics-clinical-trial-submission/ (accessed on 30 November 2022).
- Sensorion Receives US FDA Rare Pediatric Disease Designation for Gene Therapy. Available online: https://www.biopharma-reporter.com/Article/2022/11/07/sensorion-receives-us-fda-rare-pediatric-disease-designation-for-gene-therapy (accessed on 30 November 2022).



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saidia, A.R.; Ruel, J.; Bahloul, A.; Chaix, B.; Venail, F.; Wang, J. Current Advances in Gene Therapies of Genetic Auditory Neuropathy Spectrum Disorder. J. Clin. Med. 2023, 12, 738. https://doi.org/10.3390/jcm12030738
Saidia AR, Ruel J, Bahloul A, Chaix B, Venail F, Wang J. Current Advances in Gene Therapies of Genetic Auditory Neuropathy Spectrum Disorder. Journal of Clinical Medicine. 2023; 12(3):738. https://doi.org/10.3390/jcm12030738
Chicago/Turabian StyleSaidia, Anissa Rym, Jérôme Ruel, Amel Bahloul, Benjamin Chaix, Frédéric Venail, and Jing Wang. 2023. "Current Advances in Gene Therapies of Genetic Auditory Neuropathy Spectrum Disorder" Journal of Clinical Medicine 12, no. 3: 738. https://doi.org/10.3390/jcm12030738
APA StyleSaidia, A. R., Ruel, J., Bahloul, A., Chaix, B., Venail, F., & Wang, J. (2023). Current Advances in Gene Therapies of Genetic Auditory Neuropathy Spectrum Disorder. Journal of Clinical Medicine, 12(3), 738. https://doi.org/10.3390/jcm12030738