The Intrinsic Biological Identities of Iron Oxide Nanoparticles and Their Coatings: Unexplored Territory for Combinatorial Therapies



Abstract
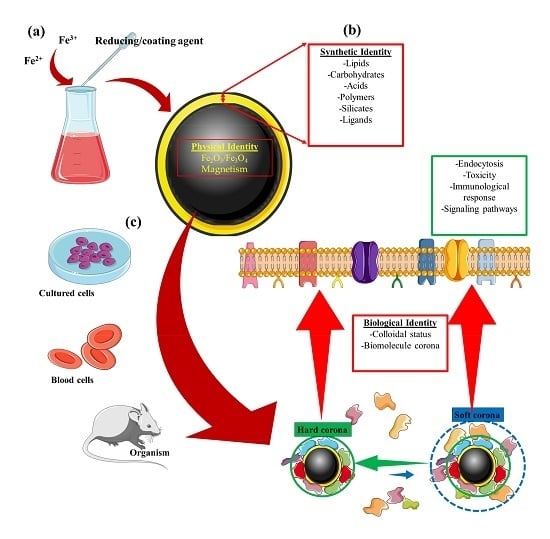
:
1. Introduction
2. Iron Oxide-Driven Biological Activities: Cellular Iron Metabolism and Reactive Oxygen Species
2.1. Cellular Components of Iron Metabolism: Macrophages
2.2. Cellular Components of Iron Metabolism: Endothelial Cells
2.3. Iron Homeostasis and Cancer Cells
2.4. Iron Oxide and Redox Homeostasis
3. Iron Oxide Nanoparticle Biodegradation
3.1. IONP Biodegradation and Biological Identity
3.2. IONP Degradation and Protein Corona
3.3. Endocytosis and IONP Degradation
3.4. IONP Biodegradation by Cellular Machinery
4. IONP Effects is Dependent on Cell Type and Status
4.1. IONPs and Myeloid Cells
4.1.1. Iron Metabolism and Macrophage Polarization
4.1.2. IONP Recognition by Macrophages and Activation
4.1.3. IONPs and Dendritic Cells (DC)
4.2. Iron Oxide and Functions of Endothelial Cells
4.3. Tumor Microenvironment and Iron Oxide Nanoparticles
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, G.; Gao, J.; Ai, H.; Chen, X. Applications and potential toxicity of magnetic iron oxide nanoparticles. Small 2013, 9, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Ling, D.; Hyeon, T. Chemical design of biocompatible iron oxide nanoparticles for medical applications. Small 2013, 9, 1450–1466. [Google Scholar] [CrossRef]
- Schladt, T.D.; Schneider, K.; Schild, H.; Tremel, W. Synthesis and bio-functionalization of magnetic nanoparticles for medical diagnosis and treatment. Dalton Trans. 2011, 40, 6315–6343. [Google Scholar] [CrossRef]
- Grover, V.P.B.; Tognarelli, J.M.; Crossey, M.M.E.; Cox, I.J.; Taylor-Robinson, S.D.; McPhail, M.J.W. Magnetic Resonance Imaging: Principles and Techniques: Lessons for Clinicians. J. Clin. Exp. Hepatol. 2015, 5, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currie, S.; Hoggard, N.; Craven, I.J.; Hadjivassiliou, M.; Wilkinson, I.D. Understanding MRI: Basic MR physics for physicians. Postgrad. Med. J. 2013, 89, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, B.; Friedman, G. Magnetic targeting for site-specific drug delivery: Applications and clinical potential. Expert Opin. Drug Deliv. 2009, 6, 53–70. [Google Scholar] [CrossRef]
- Silva, L.H.A.; Cruz, F.F.; Morales, M.M.; Weiss, D.J.; Rocco, P.R.M. Magnetic targeting as a strategy to enhance therapeutic effects of mesenchymal stromal cells. Stem Cell Res. Ther. 2017, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Kheirkhah, P.; Denyer, S.; Bhimani, A.D.; Arnone, G.D.; Esfahani, D.R.; Aguilar, T.; Zakrzewski, J.; Venugopal, I.; Habib, N.; Gallia, G.L.; et al. Magnetic Drug Targeting: A Novel Treatment for Intramedullary Spinal Cord Tumors. Sci. Rep. 2018, 8, 11417. [Google Scholar] [CrossRef]
- Price, P.M.; Mahmoud, W.E.; Al-Ghamdi, A.A.; Bronstein, L.M. Magnetic Drug Delivery: Where the Field Is Going. Front. Chem. 2018, 6, 619. [Google Scholar] [CrossRef] [Green Version]
- Mody, V.V.; Cox, A.; Shah, S.; Singh, A.; Bevins, W.; Parihar, H. Magnetic nanoparticle drug delivery systems for targeting tumor. Appl. Nanosci. 2014, 4, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.; Lim, M.; Goos, J.A.C.M.; Qiao, R.; Ng, Y.Y.; Mansfeld, F.M.; Jackson, M.; Davis, T.P.; Kavallaris, M. Biologically Targeted Magnetic Hyperthermia: Potential and Limitations. Front. Pharmacol. 2018, 9, 831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moise, S.; Byrne, J.M.; El Haj, A.J.; Telling, N.D. The potential of magnetic hyperthermia for triggering the differentiation of cancer cells. Nanoscale 2018, 10, 20519–20525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, S.L.; Walker, K.; Hoskins, C.; Telling, N.D.; Price, H.P. Nanoparticle-mediated magnetic hyperthermia is an effective method for killing the human-infective protozoan parasite Leishmania mexicana in vitro. Sci. Rep. 2019, 9, 1059. [Google Scholar] [CrossRef] [PubMed]
- Bolandparvaz, A.; Vapniarsky, N.; Harriman, R.; Alvarez, K.; Saini, J.; Zang, Z.; Van De Water, J.; Lewis, J.S. Biodistribution and toxicity of epitope-functionalized dextran iron oxide nanoparticles in a pregnant murine model. J. Biomed. Mater. Res. Part. A 2020, 108, 1186–1202. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, X.; Wang, J.; Yuan, J.; Zhang, J.; Zhu, X.; Lei, C.; Yang, Q.; Wang, B.; Cao, F.; et al. A Peptide-Functionalized Magnetic Nanoplatform-Loaded Melatonin for Targeted Amelioration of Fibrosis in Pressure Overload-Induced Cardiac Hypertrophy. Int. J. Nanomed. 2020, 15, 1321–1333. [Google Scholar] [CrossRef] [Green Version]
- Del Sol-Fernández, S.; Portilla-Tundidor, Y.; Gutiérrez, L.; Odio, O.F.; Reguera, E.; Barber, D.F.; Morales, M.P. Flower-like Mn-Doped Magnetic Nanoparticles Functionalized with αvβ3-Integrin-Ligand to Efficiently Induce Intracellular Heat after Alternating Magnetic Field Exposition, Triggering Glioma Cell Death. ACS Appl. Mater. Interfaces 2019, 11, 26648–26663. [Google Scholar] [CrossRef]
- Cędrowska, E.; Pruszyński, M.; Gawęda, W.; Żuk, M.; Krysiński, P.; Bruchertseifer, F.; Morgenstern, A.; Karageorgou, M.-A.; Bouziotis, P.; Bilewicz, A. Trastuzumab Conjugated Superparamagnetic Iron Oxide Nanoparticles Labeled with 225Ac as a Perspective Tool for Combined α-Radioimmunotherapy and Magnetic Hyperthermia of HER2-Positive Breast Cancer. Molecules 2020, 25, 1025. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Chen, S.; Li, Y.; Zeng, L.; Lian, G.; Li, J.; Chen, S.; Huang, K.; Chen, Y. Nanoparticles modified by triple single chain antibodies for MRI examination and targeted therapy in pancreatic cancer. Nanoscale 2020, 12, 4473–4490. [Google Scholar] [CrossRef] [PubMed]
- Yigit, M.V.; Mazumdar, D.; Lu, Y. MRI Detection of Thrombin with Aptamer Functionalized Superparamagnetic Iron Oxide Nanoparticles. Bioconj. Chem. 2008, 19, 412–417. [Google Scholar] [CrossRef]
- Aghanejad, A.; Babamiri, H.; Adibkia, K.; Barar, J.; Omidi, Y. Mucin-1 aptamer-armed superparamagnetic iron oxide nanoparticles for targeted delivery of doxorubicin to breast cancer cells. Bioimpacts 2018, 8, 117–127. [Google Scholar] [CrossRef]
- Yu, Q.; Xiong, X.; Zhao, L.; Xu, T.; Wang, Q. Antifibrotic effects of specific siRNA targeting connective tissue growth factor delivered by polyethyleneimine-functionalized magnetic iron oxide nanoparticles on LX-2 cells. Mol. Med. Rep. 2020, 21, 181–190. [Google Scholar] [CrossRef]
- Su, Z.; Liu, D.; Chen, L.; Zhang, J.; Ru, L.; Chen, Z.; Gao, Z.; Wang, X. CD44-Targeted Magnetic Nanoparticles Kill Head And Neck Squamous Cell Carcinoma Stem Cells In An Alternating Magnetic Field. Int. J. Nanomed. 2019, 14, 7549–7560. [Google Scholar] [CrossRef] [Green Version]
- Parak, W.J.; Gerion, D.; Pellegrino, T.; Zanchet, D.; Micheel, C.; Williams, S.C.; Boudreau, R.; Gros, M.A.L.; Larabell, C.A.; Alivisatos, A.P. Biological applications of colloidal nanocrystals. Nanotechnology 2003, 14, R15–R27. [Google Scholar] [CrossRef] [Green Version]
- Mikhaylova, M.; Kim, D.K.; Bobrysheva, N.; Osmolowsky, M.; Semenov, V.; Tsakalakos, T.; Muhammed, M. Superparamagnetism of Magnetite Nanoparticles: Dependence on Surface Modification. Langmuir 2004, 20, 2472–2477. [Google Scholar] [CrossRef] [PubMed]
- Lazarovits, J.; Chen, Y.Y.; Sykes, E.A.; Chan, W.C.W. Nanoparticle–blood interactions: The implications on solid tumour targeting. Chem. Commun. 2015, 51, 2756–2767. [Google Scholar] [CrossRef] [PubMed]
- Perrault, S.D.; Walkey, C.; Jennings, T.; Fischer, H.C.; Chan, W.C.W. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009, 9, 1909–1915. [Google Scholar] [CrossRef]
- Rezaei, G.; Daghighi, S.M.; Raoufi, M.; Esfandyari-Manesh, M.; Rahimifard, M.; Mobarakeh, V.I.; Kamalzare, S.; Ghahremani, M.H.; Atyabi, F.; Abdollahi, M.; et al. Synthetic and biological identities of polymeric nanoparticles influencing the cellular delivery: An immunological link. J. Coll. Interface Sci. 2019, 556, 476–491. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Feliu, N.; Vogt, C.; Abdelmonem, A.M.; Parak, W.J. Bridge over troubled waters: Understanding the synthetic and biological identities of engineered nanomaterials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 111–129. [Google Scholar] [CrossRef]
- Caracciolo, G.; Farokhzad, O.C.; Mahmoudi, M. Biological Identity of Nanoparticles In Vivo: Clinical Implications of the Protein Corona. Trends Biotechnol. 2017, 35, 257–264. [Google Scholar] [CrossRef]
- Albanese, A.; Walkey, C.D.; Olsen, J.B.; Guo, H.; Emili, A.; Chan, W.C.W. Secreted Biomolecules Alter the Biological Identity and Cellular Interactions of Nanoparticles. ACS Nano 2014, 8, 5515–5526. [Google Scholar] [CrossRef]
- Winn, N.C.; Volk, K.M.; Hasty, A.H. Regulation of tissue iron homeostasis: The macrophage “ferrostat”. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Klei, T.R.L.; Meinderts, S.M.; van den Berg, T.K.; van Bruggen, R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 2017, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherayil, B.J. The role of iron in the immune response to bacterial infection. Immunol. Res. 2011, 50, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philpott, C.C.; Jadhav, S. The ins and outs of iron: Escorting iron through the mammalian cytosol. Free Radic. Biol. Med. 2019, 133, 112–117. [Google Scholar] [CrossRef]
- Shokrgozar, N.; Golafshan, H.A. Molecular perspective of iron uptake, related diseases, and treatments. Blood Res. 2019, 54, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Korolnek, T.; Hamza, I. Macrophages and iron trafficking at the birth and death of red cells. Blood 2015, 125, 2893–2897. [Google Scholar] [CrossRef] [Green Version]
- Dybas, J.; Grosicki, M.; Baranska, M.; Marzec, K.M. Raman imaging of heme metabolism in situ in macrophages and Kupffer cells. Analyst 2018, 143, 3489–3498. [Google Scholar] [CrossRef]
- Youssef, L.A.; Rebbaa, A.; Pampou, S.; Weisberg, S.P.; Stockwell, B.R.; Hod, E.A.; Spitalnik, S.L. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood 2018, 131, 2581–2593. [Google Scholar] [CrossRef]
- Medina, M.V.; Sapochnik, D.; Garcia Sola, M.E.; Coso, O.A. Regulation of the Expression of Heme Oxygenase-1. Signal Transduction, Gene Promoter Activation and Beyond. Antioxid. Redox Signal. 2019. [Google Scholar] [CrossRef]
- Ryter, S.W. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch. Biochem. Biophys. 2019, 678, 108186. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wu, Y.; Tang, W. Heme Catabolic Pathway in Inflammation and Immune Disorders. Front. Pharmacol. 2019, 10, 825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recalcati, S.; Gammella, E.; Cairo, G. Ironing out Macrophage Immunometabolism. Pharmaceuticals 2019, 12, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta BBA 2009, 1790, 589–599. [Google Scholar] [CrossRef]
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell. Physiol. 2018, 233, 9179–9190. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Musci, G.; Polticelli, F.; Bonaccorsi di Patti, M.C. Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World J. Biol. Chem 2014, 5, 204–215. [Google Scholar] [CrossRef]
- Raub, T.J.; Newton, C.R. Recycling kinetics and transcytosis of transferrin in primary cultures of bovine brain microvessel endothelial cells. J. Cell. Physiol. 1991, 149, 141–151. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Kosman, D.J. Mechanistic analysis of iron accumulation by endothelial cells of the BBB. Biometals 2012, 25, 665–675. [Google Scholar] [CrossRef] [Green Version]
- Chiou, B.; Neal, E.H.; Bowman, A.B.; Lippmann, E.S.; Simpson, I.A.; Connor, J.R. Endothelial cells are critical regulators of iron transport in a model of the human blood–brain barrier. J. Cereb. Blood Flow Metab. 2018, 39, 2117–2131. [Google Scholar] [CrossRef]
- Imai, T.; Iwata, S.; Hirayama, T.; Nagasawa, H.; Nakamura, S.; Shimazawa, M.; Hara, H. Intracellular Fe2+ accumulation in endothelial cells and pericytes induces blood-brain barrier dysfunction in secondary brain injury after brain hemorrhage. Sci. Rep. 2019, 9, 6228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garton, T.; Keep, R.F.; Hua, Y.; Xi, G. Brain iron overload following intracranial haemorrhage. Stroke Vasc. Neurol. 2016, 1, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Katsu, M.; Niizuma, K.; Yoshioka, H.; Okami, N.; Sakata, H.; Chan, P.H. Hemoglobin-Induced Oxidative Stress Contributes to Matrix Metalloproteinase Activation and Blood–Brain Barrier Dysfunction in vivo. J. Cereb. Blood Flow Metab. 2010, 30, 1939–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canali, S.; Zumbrennen-Bullough, K.B.; Core, A.B.; Wang, C.-Y.; Nairz, M.; Bouley, R.; Swirski, F.K.; Babitt, J.L. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 2017, 129, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Koch, P.-S.; Olsavszky, V.; Ulbrich, F.; Sticht, C.; Demory, A.; Leibing, T.; Henzler, T.; Meyer, M.; Zierow, J.; Schneider, S.; et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 2017, 129, 415–419. [Google Scholar] [CrossRef] [Green Version]
- RICHMOND, H.G. Induction of sarcoma in the rat by iron-dextran complex. Br. Med. J. 1959, 1, 947–949. [Google Scholar] [CrossRef]
- Carter, R.L.; Mitchley, B.C.; Roe, F.J. Induction of tumours in mice and rats with ferric sodium gluconate and iron dextran glycerol glycoside. Br. J. Cancer 1968, 22, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Haddow, A.; Roe, F.J.; Mitchley, B.C. Induction of sarcomata in rabbits by intramuscular injection of iron-dextran (“imferon”). Br. Med. J. 1964, 1, 1593–1594. [Google Scholar] [CrossRef] [Green Version]
- Fielding, J. Sarcoma induction by iron-carbohydrate complexes. Br. Med. J. 1962, 1, 1800–1803. [Google Scholar] [CrossRef] [Green Version]
- Langvad, E. Iron-dextran induction of distant tumours in mice. Int. J. Cancer 1968, 3, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Akatsuka, S.; Yamashita, Y.; Ohara, H.; Liu, Y.-T.; Izumiya, M.; Abe, K.; Ochiai, M.; Jiang, L.; Nagai, H.; Okazaki, Y.; et al. Fenton reaction induced cancer in wild type rats recapitulates genomic alterations observed in human cancer. PLoS ONE 2012, 7, e43403. [Google Scholar] [CrossRef] [PubMed]
- Li, G.H.; Akatsuka, S.; Chew, S.H.; Jiang, L.; Nishiyama, T.; Sakamoto, A.; Takahashi, T.; Futakuchi, M.; Suzuki, H.; Sakumi, K.; et al. Fenton reaction-induced renal carcinogenesis in Mutyh-deficient mice exhibits less chromosomal aberrations than the rat model. Pathol. Int. 2017, 67, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Marques, O.; Porto, G.; Rêma, A.; Faria, F.; Cruz Paula, A.; Gomez-Lazaro, M.; Silva, P.; Martins da Silva, B.; Lopes, C. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer 2016, 16, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, L.D.; Coffman, L.G.; Chou, J.W.; Black, M.A.; Bergh, J.; D’Agostino, R., Jr.; Torti, S.V.; Torti, F.M. An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res. 2011, 71, 6728–6737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela, D. Iron Metabolism in Prostate Cancer; From Basic Science to New Therapeutic Strategies. Front. Oncol. 2018, 8, 547. [Google Scholar] [CrossRef]
- Zhao, B.; Li, R.; Cheng, G.; Li, Z.; Zhang, Z.; Li, J.; Zhang, G.; Bi, C.; Hu, C.; Yang, L.; et al. Role of hepcidin and iron metabolism in the onset of prostate cancer. Oncol. Lett. 2018, 15, 9953–9958. [Google Scholar] [CrossRef] [Green Version]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, Y.; Guo, W.; Yuan, L.; Zhang, D.; Xu, Y.; Nemeth, E.; Ganz, T.; Liu, S. Disordered hepcidin–ferroportin signaling promotes breast cancer growth. Cell. Signal. 2014, 26, 2539–2550. [Google Scholar] [CrossRef]
- Guo, W.; Zhang, S.; Chen, Y.; Zhang, D.; Yuan, L.; Cong, H.; Liu, S. An important role of the hepcidin–ferroportin signaling in affecting tumor growth and metastasis. Acta Biochim. Biophys. Sin. 2015, 47, 703–715. [Google Scholar] [CrossRef] [Green Version]
- Babu, K.R.; Muckenthaler, M.U. miR-20a regulates expression of the iron exporter ferroportin in lung cancer. J. Mol. Med. 2016, 94, 347–359. [Google Scholar] [CrossRef]
- Castoldi, M.; Muckenthaler, M.U. Regulation of iron homeostasis by microRNAs. Cell. Mol. Life Sci. 2012, 69, 3945–3952. [Google Scholar] [CrossRef]
- Masui, K.; Harachi, M.; Ikegami, S.; Yang, H.; Onizuka, H.; Yong, W.H.; Cloughesy, T.F.; Muragaki, Y.; Kawamata, T.; Arai, N.; et al. mTORC2 links growth factor signaling with epigenetic regulation of iron metabolism in glioblastoma. J. Biol. Chem. 2019, 294, 19740–19751. [Google Scholar] [CrossRef] [PubMed]
- Udali, S.; Castagna, A.; Corbella, M.; Ruzzenente, A.; Moruzzi, S.; Mazzi, F.; Campagnaro, T.; De Santis, D.; Franceschi, A.; Pattini, P.; et al. Hepcidin and DNA promoter methylation in hepatocellular carcinoma. Eur. J. Clin. Investig. 2018, 48, e12870. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Zhang, J.; Su, Y.; Shen, Y.-Y.; Jiang, D.-X.; Hou, Y.-Y.; Geng, M.-Y.; Ding, J.; Chen, Y. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat. Commun. 2017, 8, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, Toxicity and Oxidative Stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, K.; Brown, D.M.; Mitchell, C.; Dineva, M.; Beswick, P.H.; Gilmour, P.; MacNee, W. Free radical activity of PM10: Iron-mediated generation of hydroxyl radicals. Environ. Health Perspect. 1997, 105 (Suppl. 5), 1285–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, F.R.; He, C.; Danes, J.M.; Coelho, D.R.; Sampaio, V.P.; Gantner, B.N.; Bonini, M.G. Mitochondrial Superoxide Dismutase: What the established, the intriguing, and the novel reveal about a key cellular redox switch. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. Int. J. Mol. Sci. 2019, 20, 3673. [Google Scholar] [CrossRef] [Green Version]
- Ganguli, G.; Mukherjee, U.; Sonawane, A. Peroxisomes and Oxidative Stress: Their Implications in the Modulation of Cellular Immunity During Mycobacterial Infection. Front. Microbiol. 2019, 10, 1121. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.H.; Tran, G.-B.; Nguyen, C.T. Anti-oxidative effects of superoxide dismutase 3 on inflammatory diseases. J. Mol. Med. 2020, 98, 59–69. [Google Scholar] [CrossRef]
- Zhao, Y.; Kong, G.Y.; Pei, W.M.; Zhou, B.; Zhang, Q.Q.; Pan, B.-B. Dexmedetomidine alleviates hepatic injury via the inhibition of oxidative stress and activation of the Nrf2/HO-1 signaling pathway. Eur. Cytok. Netw. 2019, 30, 88–97. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, Y.; Miao, W.; Cheng, T. Reactive Oxygen Species and Nrf2: Functional and Transcriptional Regulators of Hematopoiesis. Oxid. Med. Cell Longev. 2019, 2019, 5153268. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Chattopadhyay, A. Nrf2–ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2020, 235, 3119–3130. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Rehmani, I.; Esaki, S.; Fu, R.; Chen, L.; de Serrano, V.; Liu, A. Pirin is an iron-dependent redox regulator of NF-κB. Proc. Natl. Acad. Sci. USA 2013, 201221743. [Google Scholar] [CrossRef] [Green Version]
- Mejías, R.; Gutiérrez, L.; Salas, G.; Pérez-Yagüe, S.; Zotes, T.M.; Lázaro, F.J.; Morales, M.P.; Barber, D.F. Long term biotransformation and toxicity of dimercaptosuccinic acid-coated magnetic nanoparticles support their use in biomedical applications. J. Control. Release 2013, 171, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, J.M.; Gavilán, H.; del Dedo, V.; Lorente-Sorolla, E.; Sanz-Ortega, L.; da Silva, G.B.; Costo, R.; Perez-Yagüe, S.; Talelli, M.; Marciello, M.; et al. Time-course assessment of the aggregation and metabolization of magnetic nanoparticles. Acta Biomater. 2017, 58, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Mazuel, F.; Espinosa, A.; Luciani, N.; Reffay, M.; Le Borgne, R.; Motte, L.; Desboeufs, K.; Michel, A.; Pellegrino, T.; Lalatonne, Y.; et al. Massive Intracellular Biodegradation of Iron Oxide Nanoparticles Evidenced Magnetically at Single-Endosome and Tissue Levels. ACS Nano 2016, 10, 7627–7638. [Google Scholar] [CrossRef] [PubMed]
- Lunov, O.; Syrovets, T.; Röcker, C.; Tron, K.; Nienhaus, G.U.; Rasche, V.; Mailänder, V.; Landfester, K.; Simmet, T. Lysosomal degradation of the carboxydextran shell of coated superparamagnetic iron oxide nanoparticles and the fate of professional phagocytes. Biomaterials 2010, 31, 9015–9022. [Google Scholar] [CrossRef] [PubMed]
- Gräfe, C.; von der Lühe, M.; Weidner, A.; Globig, P.; Clement, J.H.; Dutz, S.; Schacher, F.H. Protein corona formation and its constitutional changes on magnetic nanoparticles in serum featuring a polydehydroalanine coating: Effects of charge and incubation conditions. Nanotechnology 2019, 30, 265707. [Google Scholar] [CrossRef] [PubMed]
- Escamilla-Rivera, V.; Solorio-Rodríguez, A.; Uribe-Ramírez, M.; Lozano, O.; Lucas, S.; Chagolla-López, A.; Winkler, R.; De Vizcaya-Ruiz, A. Plasma protein adsorption on Fe(3)O(4)-PEG nanoparticles activates the complement system and induces an inflammatory response. Int. J. Nanomed. 2019, 14, 2055–2067. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Jiang, P.; Luo, B.; Lan, F.; He, J.; Wu, Y. Dynamic protein corona influences immune-modulating osteogenesis in magnetic nanoparticle (MNP)-infiltrated bone regeneration scaffolds in vivo. Nanoscale 2019, 11, 6817–6827. [Google Scholar] [CrossRef]
- Vogt, C.; Pernemalm, M.; Kohonen, P.; Laurent, S.; Hultenby, K.; Vahter, M.; Lehtiö, J.; Toprak, M.S.; Fadeel, B. Proteomics Analysis Reveals Distinct Corona Composition on Magnetic Nanoparticles with Different Surface Coatings: Implications for Interactions with Primary Human Macrophages. PLoS ONE 2015, 10, e0129008. [Google Scholar] [CrossRef] [Green Version]
- Ashby, J.; Pan, S.; Zhong, W. Size and surface functionalization of iron oxide nanoparticles influence the composition and dynamic nature of their protein corona. ACS Appl. Mater. Interfaces 2014, 6, 15412–15419. [Google Scholar] [CrossRef] [Green Version]
- Lundqvist, M.; Augustsson, C.; Lilja, M.; Lundkvist, K.; Dahlbäck, B.; Linse, S.; Cedervall, T. The nanoparticle protein corona formed in human blood or human blood fractions. PLoS ONE 2017, 12, e0175871. [Google Scholar] [CrossRef] [Green Version]
- Stepien, G.; Moros, M.; Pérez-Hernández, M.; Monge, M.; Gutiérrez, L.; Fratila, R.M.; de Las Heras, M.; Menao Guillén, S.; Puente Lanzarote, J.J.; Solans, C.; et al. Effect of Surface Chemistry and Associated Protein Corona on the Long-Term Biodegradation of Iron Oxide Nanoparticles In Vivo. ACS Appl. Mater. Interfaces 2018, 10, 4548–4560. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Bai, J.; Jiang, X. Monitoring of the Enzymatic Degradation of Protein Corona and Evaluating the Accompanying Cytotoxicity of Nanoparticles. ACS Appl. Mater. Interfaces 2015, 7, 17614–17622. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xu, P.; Ding, H.-M.; Yu, Y.-S.; Huo, D.; Ma, Y.-Q. Tailoring the component of protein corona via simple chemistry. Nat. Commun. 2019, 10, 4520. [Google Scholar] [CrossRef]
- Liu, Z.; Zhan, X.; Xu, X.; Wu, Y.; Gu, Z. Static Magnetic Field Dictates Protein Corona Formation on the Surface of Glutamine-Modified Superparamagnetic Iron Oxide Nanoparticles. Part. Part. Syst. Char. 2018, 35, 1700418. [Google Scholar] [CrossRef]
- Pombo-García, K.; Rühl, C.L.; Lam, R.; Barreto, J.A.; Ang, C.-S.; Scammells, P.J.; Comba, P.; Spiccia, L.; Graham, B.; Joshi, T.; et al. Zwitterionic Modification of Ultrasmall Iron Oxide Nanoparticles for Reduced Protein Corona Formation. Chempluschem 2017, 82, 638–646. [Google Scholar] [CrossRef]
- Ostafin, A. Nanotechnology: Nanoparticle Characterization and Application in Pharmacology and Toxicology. J. Nanomed. Nanotechnol. 2019, 10. [Google Scholar] [CrossRef]
- Mazzolini, J.; Weber, R.J.M.; Chen, H.-S.; Khan, A.; Guggenheim, E.; Shaw, R.K.; Chipman, J.K.; Viant, M.R.; Rappoport, J.Z. Protein Corona Modulates Uptake and Toxicity of Nanoceria via Clathrin-Mediated Endocytosis. Biol. Bull. 2016, 231, 40–60. [Google Scholar] [CrossRef]
- Safi, M.; Courtois, J.; Seigneuret, M.; Conjeaud, H.; Berret, J.-F. The effects of aggregation and protein corona on the cellular internalization of iron oxide nanoparticles. Biomaterials 2011, 32, 9353–9363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesniak, A.; Campbell, A.; Monopoli, M.P.; Lynch, I.; Salvati, A.; Dawson, K.A. Serum heat inactivation affects protein corona composition and nanoparticle uptake. Biomaterials 2010, 31, 9511–9518. [Google Scholar] [CrossRef]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-independent pathways of endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a016758. [Google Scholar] [CrossRef] [Green Version]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Kerr, M.C.; Teasdale, R.D. Defining macropinocytosis. Traffic 2009, 10, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flannagan, R.S.; Jaumouillé, V.; Grinstein, S. The cell biology of phagocytosis. Annu. Rev. Pathol. 2012, 7, 61–98. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Feng, Q.; Liu, Y.; Huang, J.; Chen, K.; Huang, J.; Xiao, K. Uptake, distribution, clearance, and toxicity of iron oxide nanoparticles with different sizes and coatings. Sci. Rep. 2018, 8, 2082. [Google Scholar] [CrossRef]
- Forest, V.; Pourchez, J. Preferential binding of positive nanoparticles on cell membranes is due to electrostatic interactions: A too simplistic explanation that does not take into account the nanoparticle protein corona. Mater. Sci. Eng. C 2017, 70, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Schweiger, C.; Hartmann, R.; Zhang, F.; Parak, W.J.; Kissel, T.H.; Rivera_Gil, P. Quantification of the internalization patterns of superparamagnetic iron oxide nanoparticles with opposite charge. J. Nanobiotechnol. 2012, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Mulens-Arias, V.; Rojas, J.M.; Sanz-Ortega, L.; Portilla, Y.; Pérez-Yagüe, S.; Barber, D.F. Polyethylenimine-coated superparamagnetic iron oxide nanoparticles impair in vitro and in vivo angiogenesis. Nanomedicine 2019, 21, 102063. [Google Scholar] [CrossRef]
- Bohmer, N.; Jordan, A. Caveolin-1 and CDC42 mediated endocytosis of silica-coated iron oxide nanoparticles in HeLa cells. Beilstein J. Nanotechnol. 2015, 6, 167–176. [Google Scholar] [CrossRef]
- Mulens-Arias, V.; Rojas, J.M.; Pérez-Yagüe, S.; Morales, M.P.; Barber, D.F. Polyethylenimine-coated SPIONs trigger macrophage activation through TLR-4 signaling and ROS production and modulate podosome dynamics. Biomaterials 2015, 52, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Mulens-Arias, V.; Rojas, J.M.; Pérez-Yagüe, S.; Morales, M.d.P.; Barber, D.F. Polyethylenimine-coated SPION exhibits potential intrinsic anti-metastatic properties inhibiting migration and invasion of pancreatic tumor cells. J. Control. Release 2015, 216, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gao, F.; Jiang, W.; Wu, X.; Cai, Y.; Tang, J.; Gao, X.; Gao, F. Folic acid-conjugated superparamagnetic iron oxide nanoparticles for tumor-targeting MR imaging. Drug Deliv. 2016, 23, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Petters, C.; Bulcke, F.; Thiel, K.; Bickmeyer, U.; Dringen, R. Uptake of fluorescent iron oxide nanoparticles by oligodendroglial OLN-93 cells. Neurochem. Res. 2014, 39, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Raynal, I.; Prigent, P.; Peyramaure, S.; Najid, A.; Rebuzzi, C.; Corot, C. Macrophage endocytosis of superparamagnetic iron oxide nanoparticles: Mechanisms and comparison of ferumoxides and ferumoxtran-10. Invest. Radiol. 2004, 39, 56–63. [Google Scholar] [CrossRef]
- Calero, M.; Chiappi, M.; Lazaro-Carrillo, A.; Rodríguez, M.J.; Chichón, F.J.; Crosbie-Staunton, K.; Prina-Mello, A.; Volkov, Y.; Villanueva, A.; Carrascosa, J.L. Characterization of interaction of magnetic nanoparticles with breast cancer cells. J. Nanobiotechnol. 2015, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Lunov, O.; Zablotskii, V.; Syrovets, T.; Röcker, C.; Tron, K.; Nienhaus, G.U.; Simmet, T. Modeling receptor-mediated endocytosis of polymer-functionalized iron oxide nanoparticles by human macrophages. Biomaterials 2011, 32, 547–555. [Google Scholar] [CrossRef]
- Li, Z.; Shuai, C.; Li, X.; Li, X.; Xiang, J.; Li, G. Mechanism of poly-l-lysine-modified iron oxide nanoparticles uptake into cells. J. Biomed. Mater. Res. A 2013, 101, 2846–2850. [Google Scholar] [CrossRef]
- Ayala, V.; Herrera, A.P.; Latorre-Esteves, M.; Torres-Lugo, M.; Rinaldi, C. Effect of surface charge on the colloidal stability and in vitro uptake of carboxymethyl dextran-coated iron oxide nanoparticles. J. Nanopart. Res. 2013, 15, 1874. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-J.; Gu, H.-C. Study on the endocytosis and the internalization mechanism of aminosilane-coated Fe3O4 nanoparticles in vitro. J. Mater. Sci. Mater. Med. 2007, 18, 2145–2149. [Google Scholar] [CrossRef]
- Gu, J.; Xu, H.; Han, Y.; Dai, W.; Hao, W.; Wang, C.; Gu, N.; Xu, H.; Cao, J. The internalization pathway, metabolic fate and biological effect of superparamagnetic iron oxide nanoparticles in the macrophage-like RAW264.7 cell. Sci. China Life Sci. 2011, 54, 793–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, N.L.; Estrela-Lopis, I.; Böttner, J.; Lopes, C.A.; Guido, B.C.; de Sousa, A.R.; Báo, S.N. Exploring cellular uptake of iron oxide nanoparticles associated with rhodium citrate in breast cancer cells. Int. J. Nanomed. 2017, 12, 5511–5523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cañete, M.; Soriano, J.; Villanueva, A.; Roca, A.G.; Veintemillas, S.; Serna, C.J.; Miranda, R.; Del Puerto Morales, M. The endocytic penetration mechanism of iron oxide magnetic nanoparticles with positively charged cover: A morphological approach. Int. J. Mol. Med. 2010, 26, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Maraloiu, V.-A.; Appaix, F.; Broisat, A.; Le Guellec, D.; Teodorescu, V.S.; Ghezzi, C.; van der Sanden, B.; Blanchin, M.-G. Multiscale investigation of USPIO nanoparticles in atherosclerotic plaques and their catabolism and storage in vivo. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Poller, W.C.; Pieber, M.; Boehm-Sturm, P.; Ramberger, E.; Karampelas, V.; Möller, K.; Schleicher, M.; Wiekhorst, F.; Löwa, N.; Wagner, S.; et al. Very small superparamagnetic iron oxide nanoparticles: Long-term fate and metabolic processing in atherosclerotic mice. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2575–2586. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Gutiérrez, L.; Cáceres-Vélez, P.R.; Santos, D.; Chaves, S.B.; Fascineli, M.L.; Garcia, M.P.; Azevedo, R.B.; Morales, M.P. Biotransformation of magnetic nanoparticles as a function of coating in a rat model. Nanoscale 2015, 7, 16321–16329. [Google Scholar] [CrossRef]
- Curcio, A.; Van de Walle, A.; Serrano, A.; Preveral, S.; Péchoux, C.; Pignol, D.; Menguy, N.; Lefevre, C.T.; Espinosa, A.; Wilhelm, C. Transformation Cycle of Magnetosomes in Human Stem Cells: From Degradation to Biosynthesis of Magnetic Nanoparticles Anew. ACS Nano 2019. [Google Scholar] [CrossRef]
- Zhu, L.; Pelaz, B.; Chakraborty, I.; Parak, W.J. Investigating Possible Enzymatic Degradation on Polymer Shells around Inorganic Nanoparticles. Int. J. Mol. Sci. 2019, 20, 935. [Google Scholar] [CrossRef] [Green Version]
- Sée, V.; Free, P.; Cesbron, Y.; Nativo, P.; Shaheen, U.; Rigden, D.J.; Spiller, D.G.; Fernig, D.G.; White, M.R.H.; Prior, I.A.; et al. Cathepsin L digestion of nanobioconjugates upon endocytosis. ACS Nano 2009, 3, 2461–2468. [Google Scholar] [CrossRef]
- Dukhinova, M.S.; Prilepskii, A.Y.; Shtil, A.A.; Vinogradov, V.V. Metal Oxide Nanoparticles in Therapeutic Regulation of Macrophage Functions. Nanomaterials 2019, 9, 1631. [Google Scholar] [CrossRef] [Green Version]
- Recalcati, S.; Locati, M.; Cairo, G. Systemic and cellular consequences of macrophage control of iron metabolism. Semin. Immunol. 2012, 24, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron trafficking and metabolism in macrophages: Contribution to the polarized phenotype. Trends Immunol. 2011, 32, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, M.; Wessling-Resnick, M. Iron Metabolism in the Reticuloendothelial System. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 61–88. [Google Scholar] [CrossRef]
- Ganz, T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003, 102, 783–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef]
- Zhou, Y.; Que, K.-T.; Zhang, Z.; Yi, Z.J.; Zhao, P.X.; You, Y.; Gong, J.-P.; Liu, Z.-J. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. 2018, 7, 4012–4022. [Google Scholar] [CrossRef] [Green Version]
- Agoro, R.; Taleb, M.; Quesniaux, V.F.J.; Mura, C. Cell iron status influences macrophage polarization. PLoS ONE 2018, 13, e0196921. [Google Scholar] [CrossRef] [Green Version]
- Pagani, A.; Nai, A.; Corna, G.; Bosurgi, L.; Rovere-Querini, P.; Camaschella, C.; Silvestri, L. Low hepcidin accounts for the proinflammatory status associated with iron deficiency. Blood 2011, 118, 736–746. [Google Scholar] [CrossRef]
- Hoeft, K.; Bloch, D.B.; Graw, J.A.; Malhotra, R.; Ichinose, F.; Bagchi, A. Iron Loading Exaggerates the Inflammatory Response to the Toll-like Receptor 4 Ligand Lipopolysaccharide by Altering Mitochondrial Homeostasis. Anesthesiology 2017, 127, 121–135. [Google Scholar] [CrossRef]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Invest. 2011, 121, 985–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; Passos dos Santos, R.; Gaestel, M.; David, S. TNF and Increased Intracellular Iron Alter Macrophage Polarization to a Detrimental M1 Phenotype in the Injured Spinal Cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalzon, B.; Torres, A.; Reymond, S.; Gallet, B.; Saint-Antonin, F.; Collin-Faure, V.; Moriscot, C.; Fenel, D.; Schoehn, G.; Aude-Garcia, C.; et al. Influences of Nanoparticles Characteristics on the Cellular Responses: The Example of Iron Oxide and Macrophages. Nanomaterials 2020, 10, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Zhang, Q.; Fan, S.; Zhang, A.; Liu, B.; Hong, Y.; Guo, J.; Cui, D.; Song, J. The vacuolization of macrophages induced by large amounts of inorganic nanoparticle uptake to enhance the immune response. Nanoscale 2019, 11, 22849–22859. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Y.; Liu, X.; Pan, Y.; Sun, Z.; Xue, Y.; Wang, T.; Dou, H.; Hou, Y. SPIONs enhances IL-10-producing macrophages to relieve sepsis via Cav1-Notch1/HES1-mediated autophagy. Int. J. Nanomed. 2019, 14, 6779–6797. [Google Scholar] [CrossRef] [Green Version]
- Pedro, L.; Harmer, Q.; Mayes, E.; Shields, J.D. Impact of Locally Administered Carboxydextran-Coated Super-Paramagnetic Iron Nanoparticles on Cellular Immune Function. Small 2019, 15, 1900224. [Google Scholar] [CrossRef] [Green Version]
- Jiráková, K.; Moskvin, M.; Machová Urdzíková, L.; Rössner, P.; Elzeinová, F.; Chudíčková, M.; Jirák, D.; Ziolkowska, N.; Horák, D.; Kubinová, Š.; et al. The negative effect of magnetic nanoparticles with ascorbic acid on peritoneal macrophages. Neurochem. Res. 2019. [Google Scholar] [CrossRef]
- Jin, R.; Liu, L.; Zhu, W.; Li, D.; Yang, L.; Duan, J.; Cai, Z.; Nie, Y.; Zhang, Y.; Gong, Q.; et al. Iron oxide nanoparticles promote macrophage autophagy and inflammatory response through activation of toll-like Receptor-4 signaling. Biomaterials 2019, 203, 23–30. [Google Scholar] [CrossRef]
- Zhang, L.; Tan, S.; Liu, Y.; Xie, H.; Luo, B.; Wang, J. In vitro inhibition of tumor growth by low-dose iron oxide nanoparticles activating macrophages. J. Biomater. Appl. 2019, 33, 935–945. [Google Scholar] [CrossRef]
- Liu, L.; Sha, R.; Yang, L.; Zhao, X.; Zhu, Y.; Gao, J.; Zhang, Y.; Wen, L.-P. Impact of Morphology on Iron Oxide Nanoparticles-Induced Inflammasome Activation in Macrophages. ACS Appl. Mater. Interfaces 2018, 10, 41197–41206. [Google Scholar] [CrossRef]
- Chen, S.; Chen, S.; Zeng, Y.; Lin, L.; Wu, C.; Ke, Y.; Liu, G. Size-dependent superparamagnetic iron oxide nanoparticles dictate interleukin-1β release from mouse bone marrow-derived macrophages. J. Appl. Toxicol. 2018, 38, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Liu, T.; Tang, J.; Yang, Y.; Song, H.; Tuong, Z.K.; Fu, J.; Yu, C. Mechanism of Iron Oxide-Induced Macrophage Activation: The Impact of Composition and the Underlying Signaling Pathway. J. Am. Chem. Soc. 2019, 141, 6122–6126. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.M.; Sanz-Ortega, L.; Mulens-Arias, V.; Gutiérrez, L.; Pérez-Yagüe, S.; Barber, D.F. Superparamagnetic iron oxide nanoparticle uptake alters M2 macrophage phenotype, iron metabolism, migration and invasion. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gal, J.; Zhu, H. Sequestosome 1/p62: A multi-domain protein with multi-faceted functions. Front. Biol. 2012, 7, 189–201. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 Is a Polyubiquitin Chain Binding Protein Involved in Ubiquitin Proteasome Degradation. Mol. Cell. Biol. 2004, 24, 8055. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Ni, H.-M.; Guo, F.; Ding, Y.; Shi, Y.-H.; Lahiri, P.; Fröhlich, L.F.; Rülicke, T.; Smole, C.; Schmidt, V.C.; et al. Sequestosome 1/p62 Protein Is Associated with Autophagic Removal of Excess Hepatic Endoplasmic Reticulum in Mice. J. Biol. Chem. 2016, 291, 18663–18674. [Google Scholar] [CrossRef] [Green Version]
- Villegas, M.G.; Ceballos, M.T.; Urquijo, J.; Torres, E.Y.; Ortiz-Reyes, B.L.; Arnache-Olmos, O.L.; López, M.R. Poly(acrylic acid)-Coated Iron Oxide Nanoparticles interact with mononuclear phagocytes and decrease platelet aggregation. Cell. Immunol. 2019, 338, 51–62. [Google Scholar] [CrossRef]
- Dalzon, B.; Guidetti, M.; Testemale, D.; Reymond, S.; Proux, O.; Vollaire, J.; Collin-Faure, V.; Testard, I.; Fenel, D.; Schoehn, G.; et al. Utility of macrophages in an antitumor strategy based on the vectorization of iron oxide nanoparticles. Nanoscale 2019, 11, 9341–9352. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Xue, Y.; Liu, X.; Li, Y.; Liang, H.; Dou, H.; Hou, Y. Ferumoxytol. Attenuates the Function of MDSCs to Ameliorate LPS-Induced Immunosuppression in Sepsis. Nanoscale Res. Lett. 2019, 14, 379. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Sarkar, S.; Korchinski, D.J.; Wu, Y.; Yong, V.W.; Dunn, J.F. MRI monitoring of monocytes to detect immune stimulating treatment response in brain tumor. Neuro. Oncol. 2017, 19, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Xie, L.; Long, X.; Wang, Z.; He, C.-Y.; Chen, Z.-Y.; Zhang, L.; Nan, X.; Lei, H.; Liu, X.; et al. Efficacy of MRI visible iron oxide nanoparticles in delivering minicircle DNA into liver via intrabiliary infusion. Biomaterials 2013, 34, 3688–3696. [Google Scholar] [CrossRef] [PubMed]
- Zini, C.; Venneri, M.A.; Miglietta, S.; Caruso, D.; Porta, N.; Isidori, A.M.; Fiore, D.; Gianfrilli, D.; Petrozza, V.; Laghi, A. USPIO-labeling in M1 and M2-polarized macrophages: An in vitro study using a clinical magnetic resonance scanner. J. Cell. Physiol. 2018, 233, 5823–5828. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, Z.; Xue, Y.; Wang, G.; Cheng, Y.; Pan, Y.; Zhao, S.; Hou, Y. Anti-tumor macrophages activated by ferumoxytol. combined or surface-functionalized with the TLR3 agonist poly (I: C) promote melanoma regression. Theranostics 2018, 8, 6307–6321. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Serkova, N.J.; Groman, E.V.; Scheinman, R.I.; Simberg, D. Feraheme (Ferumoxytol) Is Recognized by Proinflammatory and Anti-inflammatory Macrophages via Scavenger Receptor Type AI/II. Mol. Pharmaceut. 2019, 16, 4274–4281. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, J.; Zhang, M.; Wang, Q.; Chen, B.; Hou, Y.; Lu, K. Ferumoxytol. and CpG oligodeoxynucleotide 2395 synergistically enhance antitumor activity of macrophages against NSCLC with EGFR(L858R/T790M) mutation. Int. J. Nanomed. 2019, 14, 4503–4515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-X.; Zhang, Y.; Dong, X.; Zhang, L.; Liu, M.-D.; Li, B.; Zhang, M.-K.; Feng, J.; Zhang, X.-Z. Artificially Reprogrammed Macrophages as Tumor-Tropic Immunosuppression-Resistant Biologics to Realize Therapeutics Production and Immune Activation. Adv. Mater. 2019, 31, 1807211. [Google Scholar] [CrossRef]
- Hao, N.-B.; Lü, M.-H.; Fan, Y.-H.; Cao, Y.-L.; Zhang, Z.-R.; Yang, S.-M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Iqbal, M.Z.; Liu, C.; Xing, J.; Akakuru, O.U.; Fang, Q.; Li, Z.; Dai, Y.; Li, A.; Guan, Y.; et al. Engineered nano-immunopotentiators efficiently promote cancer immunotherapy for inhibiting and preventing lung metastasis of melanoma. Biomaterials 2019, 223, 119464. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.; Zhu, W.; Yang, L.; Liu, L.; Jin, R.; Duan, J.; Anderson, J.M.; Ai, H. Lactosylated N-Alkyl polyethylenimine coated iron oxide nanoparticles induced autophagy in mouse dendritic cells. Regen. Biomater. 2018, 5, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Dong, H.; Zhou, N.; Dong, S.; Chen, L.; Zhu, Y.; Hu, H.-M.; Mou, Y. SPIO Enhance the Cross-Presentation and Migration of DCs and Anionic SPIO Influence the Nanoadjuvant Effects Related to Interleukin-1β. Nanoscale Res. Lett. 2018, 13, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhao, X.; Cheng, Y.; Guo, X.; Yuan, W. Iron Oxide Nanoparticles-Based Vaccine Delivery for Cancer Treatment. Mol. Pharmaceut. 2018, 15, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-G.; Zhang, Y.-L.; Zhou, Q.-Q.; Wang, X.-H.; Zhan, L.-S. Impairment of mitochondrial dynamics involved in iron oxide nanoparticle-induced dysfunction of dendritic cells was alleviated by autophagy inhibitor 3-methyladenine. J. Appl. Toxicol. 2020. [Google Scholar] [CrossRef]
- Duan, J.; Du, J.; Jin, R.; Zhu, W.; Liu, L.; Yang, L.; Li, M.; Gong, Q.; Song, B.; Anderson, J.M.; et al. Iron oxide nanoparticles promote vascular endothelial cells survival from oxidative stress by enhancement of autophagy. Regen. Biomater. 2019, 6, 221–229. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Miao, Y.; Chen, Z.; Qiang, P.; Cui, L.; Jing, H.; Guo, Y. Magnetic ferroferric oxide nanoparticles induce vascular endothelial cell dysfunction and inflammation by disturbing autophagy. J. Hazard. Mater. 2016, 304, 186–195. [Google Scholar] [CrossRef]
- Wen, T.; Du, L.; Chen, B.; Yan, D.; Yang, A.; Liu, J.; Gu, N.; Meng, J.; Xu, H. Iron oxide nanoparticles induce reversible endothelial-to-mesenchymal transition in vascular endothelial cells at acutely non-cytotoxic concentrations. Part. Fibre Toxicol. 2019, 16, 30. [Google Scholar] [CrossRef] [Green Version]
- Astanina, K.; Simon, Y.; Cavelius, C.; Petry, S.; Kraegeloh, A.; Kiemer, A.K. Superparamagnetic iron oxide nanoparticles impair endothelial integrity and inhibit nitric oxide production. Acta Biomater. 2014, 10, 4896–4911. [Google Scholar] [CrossRef]
- Matuszak, J.; Dörfler, P.; Zaloga, J.; Unterweger, H.; Lyer, S.; Dietel, B.; Alexiou, C.; Cicha, I. Shell matters: Magnetic targeting of SPIONs and in vitro effects on endothelial and monocytic cell function. Clin. Hemorheol. Microcirc. 2015, 61, 259–277. [Google Scholar] [CrossRef]
- Xia, S.; Lal, B.; Tung, B.; Wang, S.; Goodwin, C.R.; Laterra, J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro. Oncol. 2016, 18, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-J.; Lei, K.-F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jiang, Y.-C.; Sun, C.-K.; Chen, Q.-M. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep. 2016, 35, 2499–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanmee, T.; Ontong, P.; Itano, N. Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett. 2016, 375, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Lissat, A.; Joerschke, M.; Shinde, D.A.; Braunschweig, T.; Meier, A.; Makowska, A.; Bortnick, R.; Henneke, P.; Herget, G.; Gorr, T.A.; et al. IL6 secreted by Ewing sarcoma tumor microenvironment confers anti-apoptotic and cell-disseminating paracrine responses in Ewing sarcoma cells. BMC Cancer 2015, 15, 552. [Google Scholar] [CrossRef] [PubMed]
- Passi, A.; Vigetti, D.; Buraschi, S.; Iozzo, R.V. Dissecting the role of hyaluronan synthases in the tumor microenvironment. FEBS J. 2019, 286, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell 2017, 65, 730–742.e5. [Google Scholar] [CrossRef] [Green Version]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Felici, C.; Stucci, L.S.; Cives, M.; Silvestris, F. Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front. Oncol. 2019, 9, 1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa da Silva, M.; Breckwoldt, M.O.; Vinchi, F.; Correia, M.P.; Stojanovic, A.; Thielmann, C.M.; Meister, M.; Muley, T.; Warth, A.; Platten, M.; et al. Iron Induces Anti-tumor Activity in Tumor-Associated Macrophages. Front. Immunol. 2017, 8, 1479. [Google Scholar] [CrossRef] [Green Version]
- Reichel, D.; Tripathi, M.; Perez, J.M. Biological Effects of Nanoparticles on Macrophage Polarization in the Tumor Microenvironment. Nanotheranostics 2019, 3, 66–88. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-J.; Kim, J.-J.; Kang, K.-Y.; Paik, M.-J.; Lee, G.; Yee, S.-T. Enhanced anti-tumor immunotherapy by silica-coated magnetic nanoparticles conjugated with ovalbumin. Int. J. Nanomed. 2019, 14, 8235–8249. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
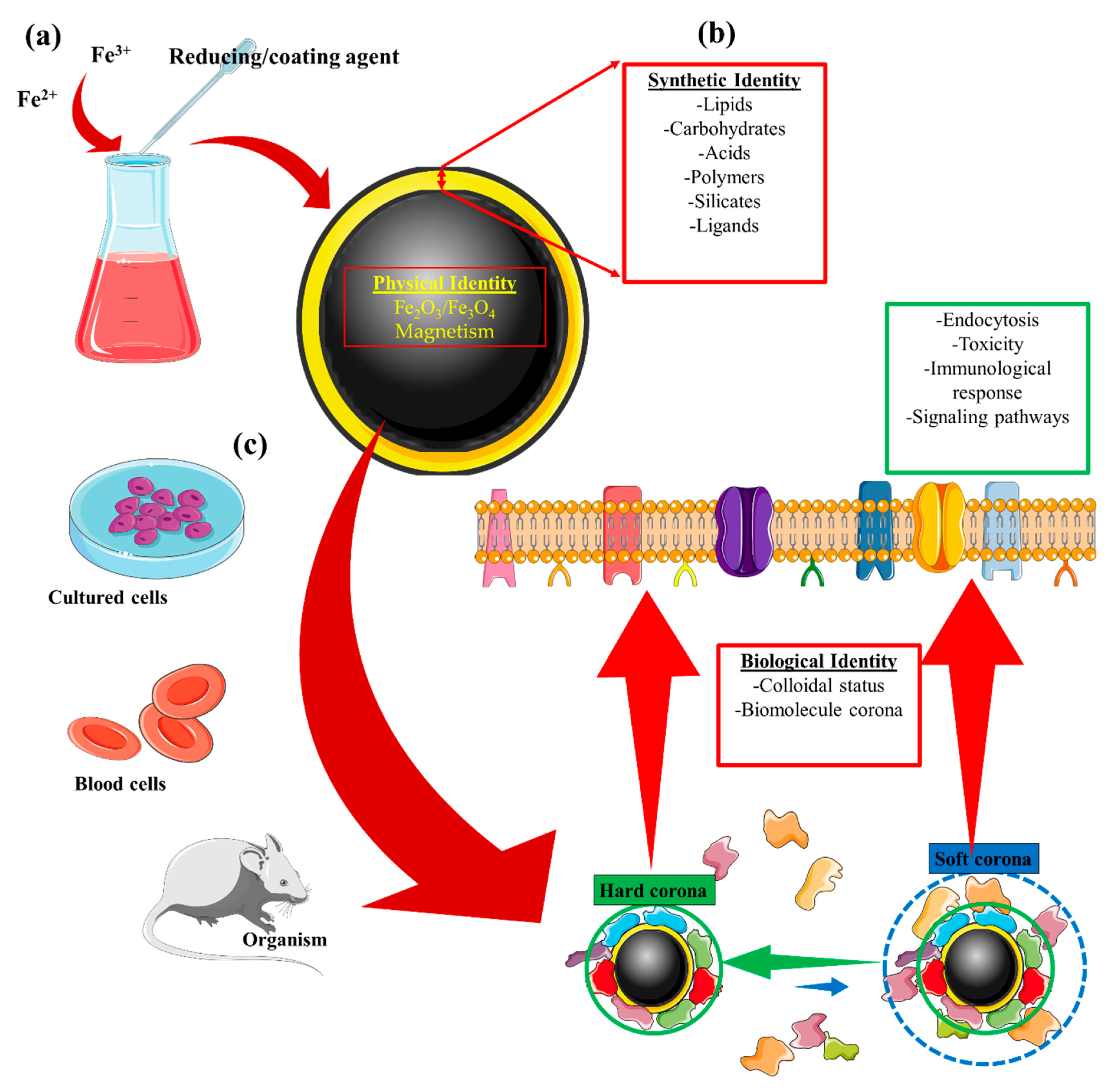{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identity | Concept |
|---|---|
| Physical | This refers to the basic physical properties that define the nanoparticle core, e.g., superparamagnetism, plasmonic, or fluorescence [23,24]. |
| Synthetic | Refers to the intrinsic physicochemical properties of the engineered surface coating, as well as its size, shape, and surface chemistry post-synthesis (surface coating modifications) [25,26,27,28]. |
| Biological | Refers to the size and aggregation state of the nanoparticles in physiological fluids (i.e., blood, tissue micro-environment, intracellular space) and the biomolecule (e.g., protein) corona. Biological identity varies with changes in synthetic identity, microenvironment, and interaction time [27,28,29,30]. |
| Iron Oxide Nanoparticle | Physical Identity | Synthetic Identity (Surface Coating) | Biological Fluid | Biological Identity |
|---|---|---|---|---|
| IONP@Glu [100] | Fe3O4 | Poly(maleic anhydride-alt-1-octadecene)-EDC-glucose | Serum protein | Protein AMBP Coagulation factor XI Fibrinogen beta chain C4b-binding protein α-like Profilin-1 |
| IONP@PEG [100] | Fe3O4 | Poly(maleic anhydride-alt-1-octadecene)-EDC-PEG | Serum protein | Actin, aortic smooth muscle Keratin, type I cytoskeletal 10 Keratin, type II cytoskeletal 7 Lysozyme C Fructose-biphosphate aldolase |
| IONP@PMAO [100] | Fe3O4 | Poly(maleic anhydride-alt-1-octadecene) | Serum protein | Fibrinogen α-chain Tubulin α-4A chain Adenylyl cyclase-associated protein Macrophage migratory inhibitory factor Ectonucleotide Pyrophosphatase/phosphodiesterase family Member 2 |
| ZW-L1@PAA-USPIONs [104] | Fe3O4 | ZW-L1@PAA | 80% human serum | α-2-Macroglobulin precursor Apolipoprotein C-II precursor |
| ZW-L2@PAA-USPIONs [104] | Fe3O4 | ZW-L2@PAA | 80% human serum | Apolipoprotein C-II precursor Apolipoprotein A-I preprotein Apolipoprotein A-II preprotein Apolipoprotein A-IV precursor Serum albumin preprotein |
| ZW-L3@PAA-USPIONs [104] | Fe3O4 | ZW-L3@PAA | 80% human serum | Apolipoprotein B-100 precursor Vitronectin precursor Complement C3 precursor Serum albumin preprotein Apolipoprotein A-II preprotein |
| PAA@USPIONs [104] | Fe3O4 | PAA | 80% human serum | α-2-Macroglobulin precursor Apolipoprotein A-I preprotein Apolipoprotein C-III precursor Complement C3 precursor Apolipoprotein B-100 precursor |
| Rh-Citrate@ IONPs [105] | Fe3O4 | Rhodium citrate | Human blood serum | Human serum albumin Complement C5 A-Kinase anchor protein 13 Apolipoprotein A-I α-2-HS-glycoprotein |
| @IONPs/PVP @IONPs/PEG @IONPs [95] | Fe3O4 | Polyvinylpyrrolidone (PVA) or polyethylene glycol (PEG) | Human plasma | 14-3-3-Protein β/α 14-3-3-Protein ε Protein kinase C inhibitor protein 1 78 kDa glucose-regulated protein (GRP-78) Actin, aortic smooth muscle (α-actin-2) |
| CSNP [97] | Fe3O4 | Silica | Human plasma | Fibrinogen b Fibrinogen g Fibrinogen a Vitronectin Histidine-rich glycoprotein |
| Nanomag-D@SPIO [97] | Fe3O4 | Dextran | Human plasma | Kininogen 1 microtubule-associated ser/thr Kinase-like Actin, beta Integrin, alpha 2b Pro-platelet basic protein |
| IONP Name | Physical Identity | Synthetic Identity (Surface Coating) | Biological Identity | Endocytic Pathways | Receptors | Cell Type | References |
|---|---|---|---|---|---|---|---|
| Silica@IONP | Fe2O3 | SiO2 | N.A. | Caveolin-dependent | CDC42 | HeLa | [120] |
| PEI@SPIONs | Fe3O4 | Polyethyleneimine (PEI) | N.A. | Clathrin-dependent and caveolin-dependent | TLR4 | RAW264.7 and Pan02 | [121,122] |
| FA–PEI@SPIONs | Fe3O4 | PEI | Folic acid | Clathrin-dependent | Folic acid receptor | HeLa | [123] |
| BP-D@IONPs | Fe2O3 | DMSA and BODIPY | With/out 10% serum (aggregates) | Endocytosis-independent and clathrin-dependent | N.A. | Oligodendroglial (OLN-93) | [124] |
| Ferumoxides | Fe3O4 | Dextran | N.A. | Clathrin-dependent | SR-A | THP-1 | [125] |
| DMSA@SPIONs | Fe3O4 | DMSA | N.A. | Clathrin-dependent (<200 nm) and macropinocytosis (aggregates > 200 nm) | N.A. | MCF-7 | [126] |
| Carboxydextran@USPION | Fe3O4 | Carboxydextran | N.A. | Clathrin-dependent | SR-A | Human macrophages | [127] |
| PLL@IONPs | N.A. | Poly-L-lysine | N.A. | Clathrin-dependent | TfR | HeLa | [128] |
| Carboxymethyl-dextran@IONPs | N.A | Carboxymethyl-dextran | Serum (protein corona) | Clathrin-dependent and caveolin-dependent | N.A. | CaCo-2 | [129] |
| Aminosilane@IONPs | Fe3O4 | Aminosilane | N.A. | Phagocytosis | N.A. | Lung cancer cell, SPC-A1 | [130] |
| DMSA@IONPs | γ-Fe2O3 | DMSA | N.A. | Clathrin-dependent, caveolin-dependent, and macropinocytosis | N.A. | RAW264.7 | [131] |
| Maghemite–rhodium citrate NPs | γ-Fe2O3 | Rh-citrate | N.A. | Clathrin-dependent | N.A. | MCF-7, MDA-MB-231, and HNTMCs | [132] |
| Aminodextran@IONPs | Fe3O4 | Aminodextran | N.A. | Macropinocytosis | N.A. | A-549 | [133] |
| PEI@IONPs | Fe3O4 | PEI | N.A. | Adsorptive endocytosis | N.A. | RAW264.7 | [116] |
| PEG@IONPs | Fe3O4 | Polyethylene glycol | N.A. | Receptor-mediated endocytosis | N.A. | RAW264.7 | [116] |
| Iron Oxide Nanoparticles | Physical Identity | Synthetic Identity (Surface Coating) | Cell Type | Described Effects | Mechanism |
|---|---|---|---|---|---|
| Carboxymaltose@Fe2O3 [153] | Fe2O3 | Carboxymaltose | J774A.1 Primary macrophages | Inhibits LPS-induced NO Inhibits IL-6 and TNFα secretion Hampers phagocytosis | Decreased free glutathione |
| PMA@IONPs (4 and 14 nm) [154] | Fe3O4 | PMA | Hamper cell viability Promote extensive vacuolization Induce TNFα, CD86 and inhibit CD206 gene expression | Promotion of extensive vacuolization | |
| PEGylated PMA@IONPs (4 and 14 nm) [154] | Fe3O4 | PEGylates PMA | RAW264.7 | Promote cell proliferation Promote extensive vacuolization Induce TNFα, CD86 and inhibit CD206 gene expression | Promotion of extensive vacuolization |
| PDSCE@IONPs [155] | γ-Fe2O3 | Polydextrose sorbitol carboxymethyl-ether | In vivo and RAW264.7 | Reduce the level of LPS-induced injury Induce a large amount of IL-10 Trigger autophagy | Promotion of autophagy through Cav1-Notch1/HES1 |
| Carboxydextran@IONPs [156] | Fe3O4 | Carboxydextran | In vivo local administration and J774.2 | Downmodulate CD86, MHC-II, Arg1 and CD163 expression (transient) Hamper phagocytosis (transient) | N.A. |
| SiO2@IONPs [157] | γ-Fe2O3 | SiO2 | Peritoneal macrophages | Increase γH2AX (marker for double-strand break) Increase IL-10 production | N.A. |
| Resovist [158] | Fe3O4 γ-Fe2O3 | Carboxydextran | Primary macrophages and RAW264.7 | Induce autophagy Induce pro-inflammatory gene expression (TNFα, IL-12, MIP-1-α, etc.) | Promote autophagy through TLR4-p38-Nrf2-p62 signaling pathway |
| Feraheme [158] | Fe3O4 | Carboxymethyl dextran | Induce autophagy Induce pro-inflammatory gene expression (TNFα, IL-12, MIP-1-α, etc.) | Promote autophagy through TLR4-p38-Nrf2-p62 signaling pathway | |
| DMSA@IONPs [159] | Fe3O4 | DMSA | RAW264.7 | Induce pro-inflammatory cytokines Promote cell proliferation Promote macrophage migration Promote macrophage-driven Hepa1-6 cell killing | N.A. |
| PEI@IONPs [121] | Fe3O4 γ-Fe2O3 | PEI | RAW264.7, THP-1, and primary peritoneal macrophages | Induce pro-inflammatory cytokines (IL-12, IL-1β, TNFα, etc.) Activate macrophages (increase CD40, CD80, CD86 and I-A/I-E) Activate the MAPK-dependent pathway Promote podosome formation and reduce ECM degradation | At least part of the effects are mediated by production of ROS and activation of TLR-4 |
| Citrate@Fe3O4 of different shape (octopod, plate, cube, sphere) [160] | Mn-doped Fe3O4 | Citrate | Bone marrow-derived macrophages (BMDMs) | Activate inflammasome (IL-1β) Induce pyroptosis Induce ROS production In this order: Octopod > plate > cube > sphere | Lysosome damage, ROS production, and K+ efflux, partially mediated by NLPR3 |
| Alkyl-PEI@IONPs (30, 80, and 120 nm) [161] | Fe3O4 | Alkyl-PEI | BMDMs | Induce IL-1β nm > 80 nm > 120 nm) Lysosome damage ROS production | Modulated by ROS |
| Fe2O3@D-SiO2 [162] | Fe2O3 | SiO2 | RAW264.7 | Increase CD80, CD86 and CD64 | Activate NF-κB and IRF5 |
| Fe3O4@D-SiO2 [162] | Fe3O4 | SiO2 | RAW264.7 | Negligible effect | N.A. |
| DMSA@IONPs [163] | Fe3O4 | DMSA | M2-like THP1 BMDMs (M2) | Induce ROS production Change Fe metabolism to an iron-replete status Reduce Mac3, CD80 Increase IL-10 production Decrease migration but increase invasion | Activation of MAPK signaling |
| APS@IONPs [163] | Fe3O4 | 3-Aminopropyl triethoxysilane | M2-like THP1 BMDMs (M2) | Induce ROS production Change Fe metabolism to an iron-replete status Reduce Mac3, CD80 Increase IL-10 production Decrease migration but increase invasion | Activation of MAPK signaling |
| AD@IONPs [163] | Fe3O4 | Aminodextran | M2-like THP1 BMDMs (M2) | Induce ROS production Change Fe metabolism to an iron-replete status Reduce Mac3 Decrease migration but increase invasion | Activation of MAPK signaling |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mulens-Arias, V.; Rojas, J.M.; Barber, D.F. The Intrinsic Biological Identities of Iron Oxide Nanoparticles and Their Coatings: Unexplored Territory for Combinatorial Therapies. Nanomaterials 2020, 10, 837. https://doi.org/10.3390/nano10050837
Mulens-Arias V, Rojas JM, Barber DF. The Intrinsic Biological Identities of Iron Oxide Nanoparticles and Their Coatings: Unexplored Territory for Combinatorial Therapies. Nanomaterials. 2020; 10(5):837. https://doi.org/10.3390/nano10050837
Chicago/Turabian StyleMulens-Arias, Vladimir, José Manuel Rojas, and Domingo F. Barber. 2020. "The Intrinsic Biological Identities of Iron Oxide Nanoparticles and Their Coatings: Unexplored Territory for Combinatorial Therapies" Nanomaterials 10, no. 5: 837. https://doi.org/10.3390/nano10050837
APA StyleMulens-Arias, V., Rojas, J. M., & Barber, D. F. (2020). The Intrinsic Biological Identities of Iron Oxide Nanoparticles and Their Coatings: Unexplored Territory for Combinatorial Therapies. Nanomaterials, 10(5), 837. https://doi.org/10.3390/nano10050837