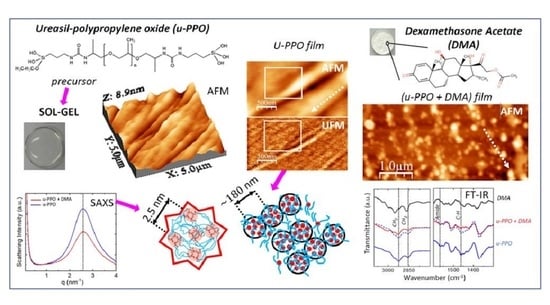
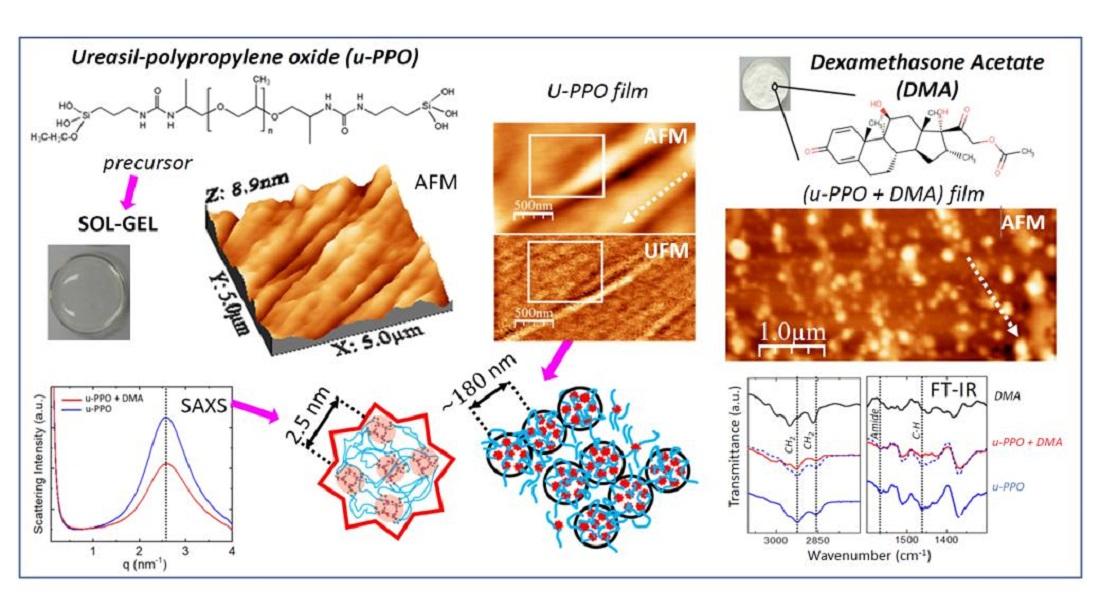
3. Results and Discussion
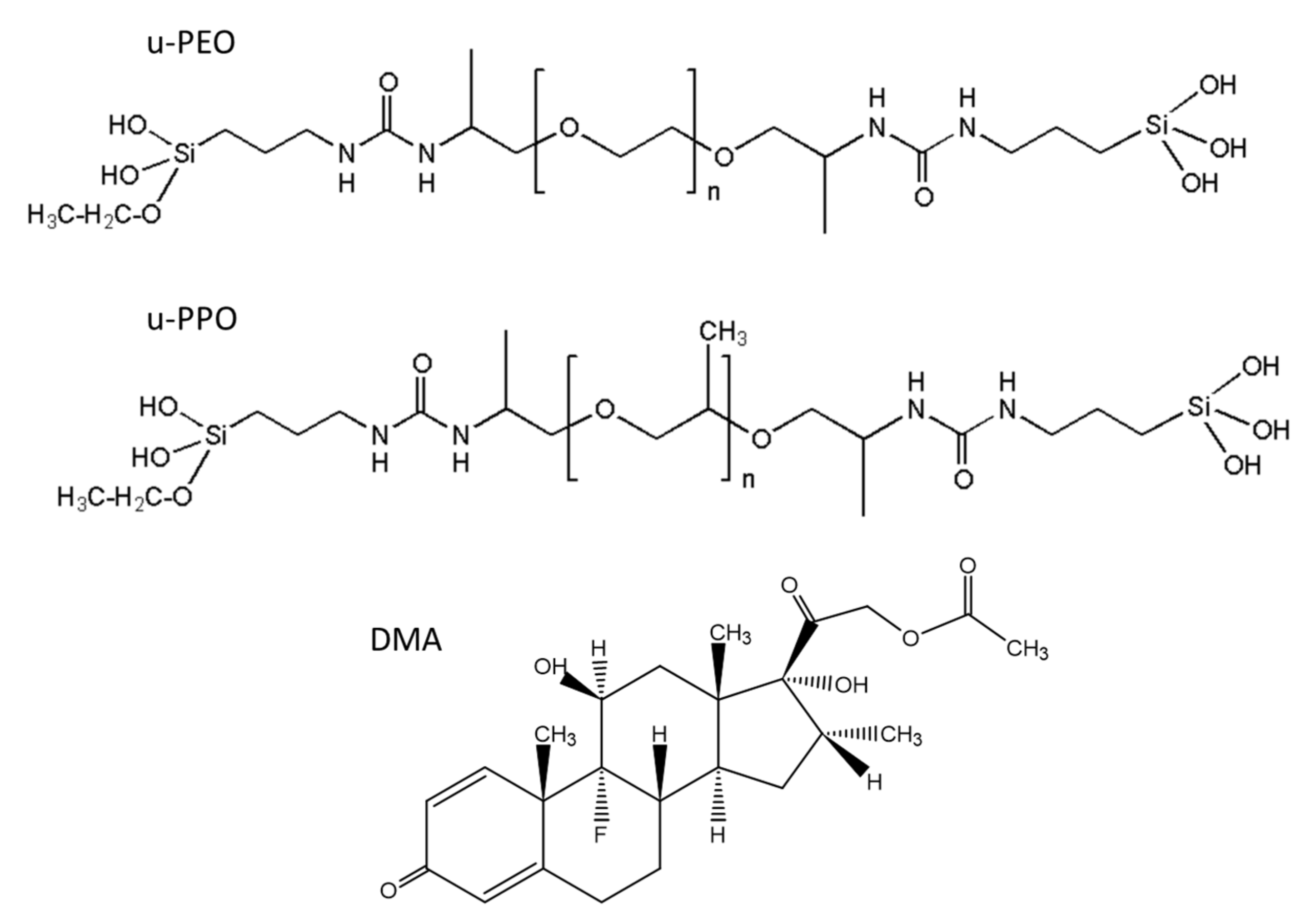
Figure 1 shows the structural formulas of the precursor molecules of u-PEO and u-PPO, and DMA. For u-PEO500, the PEO chain length contains less than n = 12 oxyethylene units, and for u-PPO400, the PPO chain length contains less than n = 7 oxypolypropylene units. As hydrolysis and condensation reactions take place during the sol-gel process, the silanol terminal groups of different molecules interact with each other to form the inorganic siloxane nodes that create the matrix network. Besides, interactions among the urea and polyether moieties from different molecules may also occur. In particular, in low molecular weight ureasil-polyether like ours, the number of urea-urea linkages is expected to be quite large [
4]. The formation of hydrogen-bonded urea-polyether associations is also possible, as the N-H groups of the urethane linkages are donor sites, and the ether oxygens, hydrogen bond acceptors. As the DMA molecules dissolve together with the precursor molecules when preparing the films, they may also interact and/or influence the sol-gel reactions leading to film formation [
13].
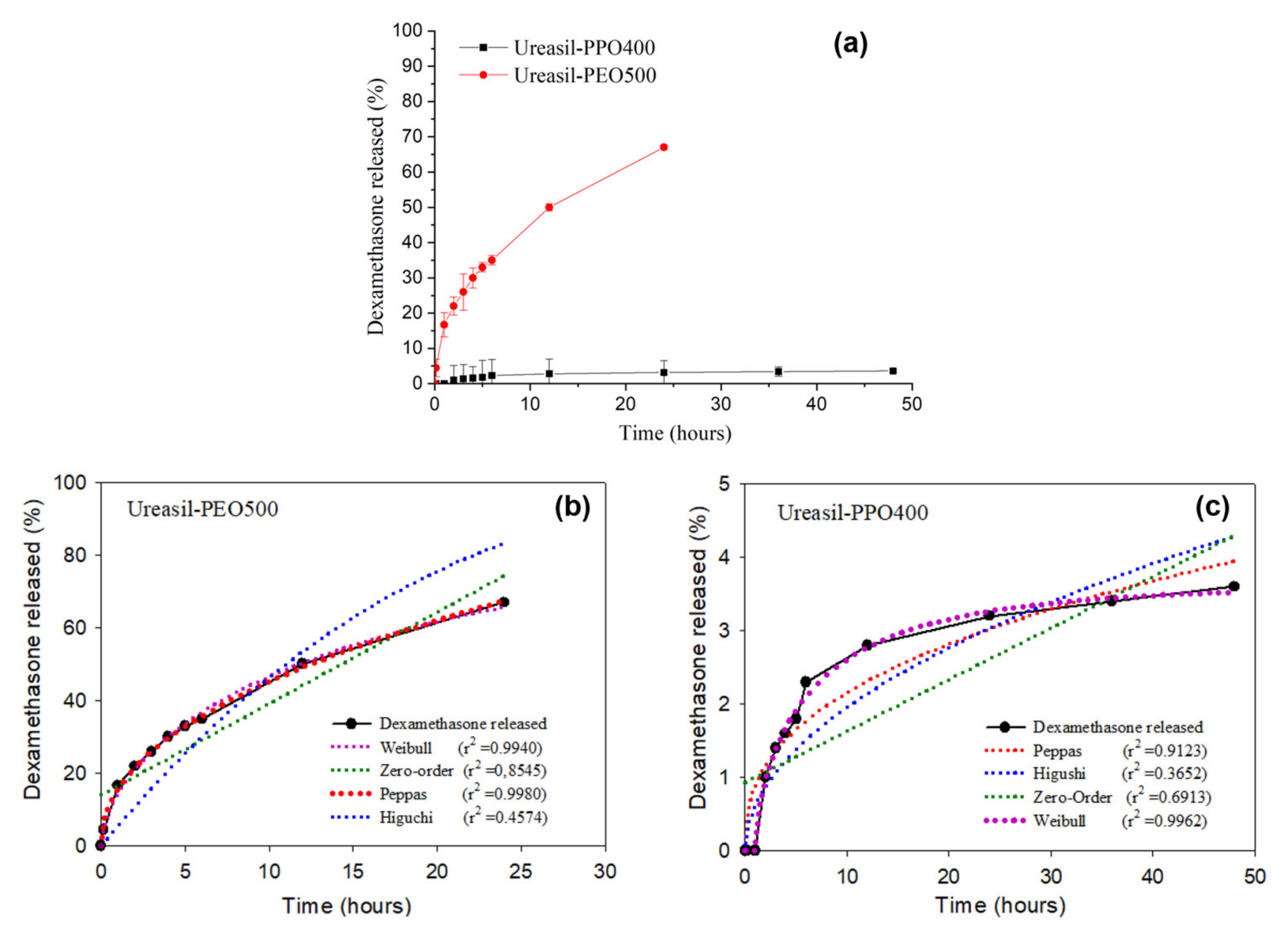
Figure 2 presents the release profiles of DMA-loaded u-PEO500 and u-PPO400 films.
Figure 2a allows us to visualize the different release rates of the films.
Figure 2b,c show the different mathematical models applied to fit the DMA release profile in each case.
As it is clearly noticeable from
Figure 2a, the samples prepared with u-PEO500 exhibit much faster release rates than those prepared with u-PPO400. Typically, release from u-PEO matrices is much faster than from u-PPO matrices, because in hydrophilic u-PEO the drug molecules can easily diffuse into the release medium through the free volume of the swollen network [
1,
2]. Whereas u-PEO has a highly hydrophilic character, the presence of the additional methyl group in u-PPO decreases its hydrophilicity. As a result, its affinity for the dissolution medium is decreased, and a lower relaxation of the polymer chains and a lower degree of swelling of the u-PPO matrix is expected. In our case, u-PEO500 and u-PPO400 hybrid materials have similar molecular weight, so the molecular weight of the polymer chain is not a factor in determining the different release profiles.
Fitting drug release data using mathematical models provides a tool to elucidate the main transport mechanisms that control the drug release process [
20,
24,
25,
26,
27]. In
Figure 2b,c Higuchi [
28], Peppas [
29] and Weibull [
30] models have been considered. The criterion used to choose among these models is the statistical coefficient of determination (r
2), which is used to evaluate the fit of the model equation. According to the values of r
2, the DMA release from u-PEO500 (
Figure 2b) fits best with the Peppas model, while that of u-PPO400 (
Figure 2c) fits best with the Weibull model.
The Peppas model is based on a power law correlation between drug release and time (
). For the Peppas model the values of exponent n determine the mechanism of drug transport out of matrix [
29]. When values of
n are less than 0.45, release is expected to occur by Fickian diffusion, whereas values of n between 0.45 ˂
n ˂ 0.89 suggest that the release is governed by anomalous transport, involving both matrix swelling and drug diffusion. In a swelling-controlled release mechanism, release depends mostly on solvent penetration. For u-PEO500, the value of the exponent
n is 0.4574, indicating that the release of the drug from the matrix to the medium presumably occurs by anomalous transport.
The Weibull model is an empirical approach, not based in any kinetic theory
. Nevertheless, reports in the literature indicate that the Weibul model does provide information about the drug release process, with the value of the exponent b correlated with the mechanism of drug transport out of the matrix [
30]. When the value of
b is less than 0.75, release is expected to occur by Fickian diffusion. The u-PPO presented a b exponent value of 0.7409. Therefore, the release of DMA from DMA-loaded u-PPO400 presumably occurs mostly by Fickian diffusion.
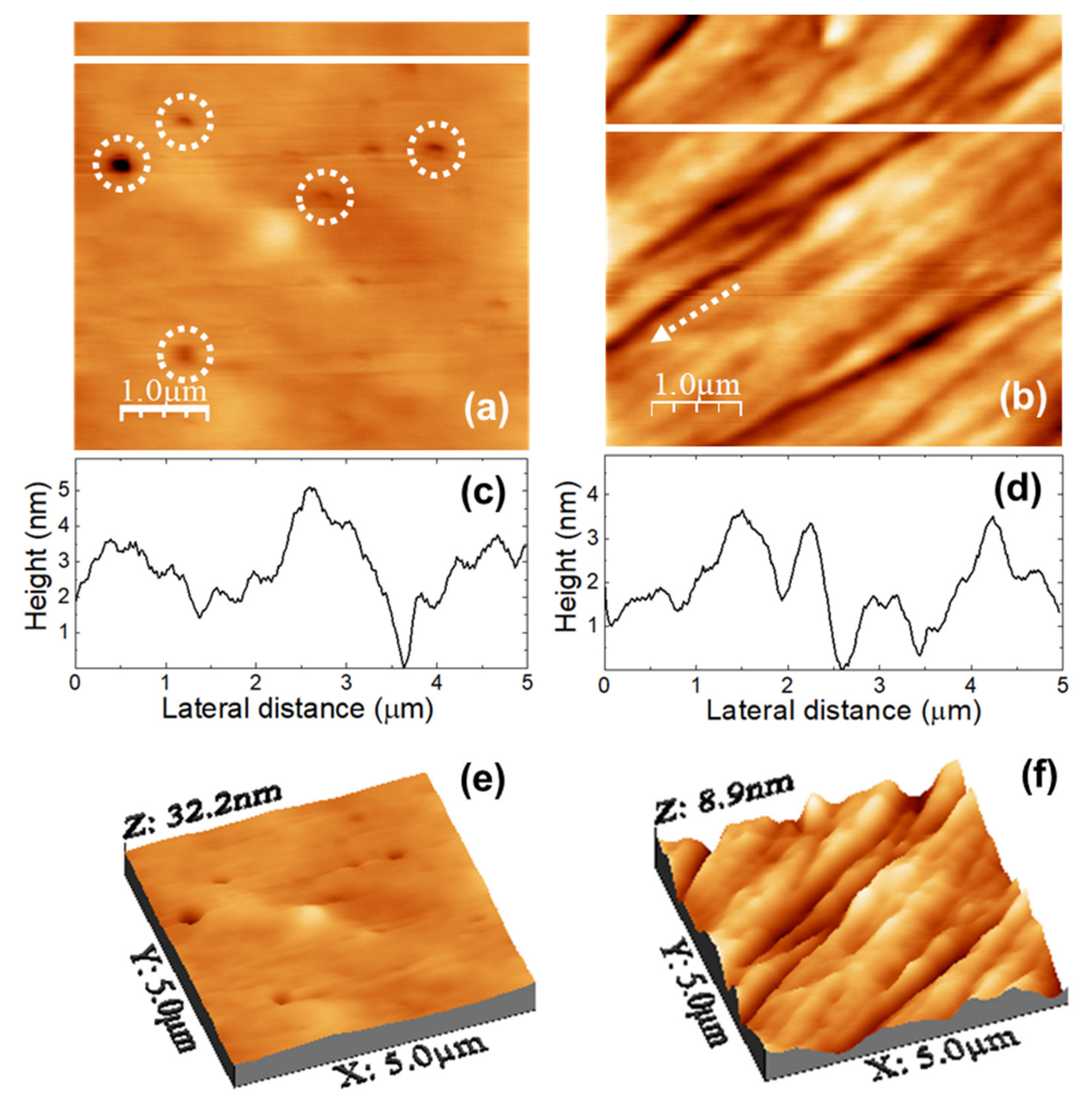
Figure 3 shows the topography of (5000 × 5000) nm surface areas recorded on unloaded (a) u-POE500 and (b) u-PPO400 films using contact-mode AFM.
Figure 3c,d correspond to height contour profiles along the continuous white lines in
Figure 3a,b, respectively.
Figure 3e,f are 3D representations of
Figure 3a,b. The surface of the u-PEO500 film is characterized by scattered pores as large as ≈200 nm in diameter; some of the pores have been pointed out with dashed white circles in
Figure 3a, and are apparent in
Figure 3e. On the u-PPO400 films’ surfaces polymer “ropes” are apparent (see
Figure 3b,f) oriented along a well-defined direction, indicated by a dashed white arrow in
Figure 3b. The root mean square (RMS) roughness on
Figure 3a (u-PEO500) and
Figure 3b (u-PPO400) is of 1.80 nm and 1.35 nm, respectively. The presence of pores causes the colour-scale range in the u-PEO image to be much larger than in the u-PPO image (see
Figure 3e,f). Nevertheless, in the areas where there are no pores, the surface corrugation is only slightly larger on the u-PEO than on the u-PPO surface, as can be seen in
Figure 3c,d. The pores favor the penetration of water into the matrix, leading to swelling, and thus modifying the surface morphology. The presence of pores helps to explain the higher drug release rates in u-PEO than in u-PPO films [
31].
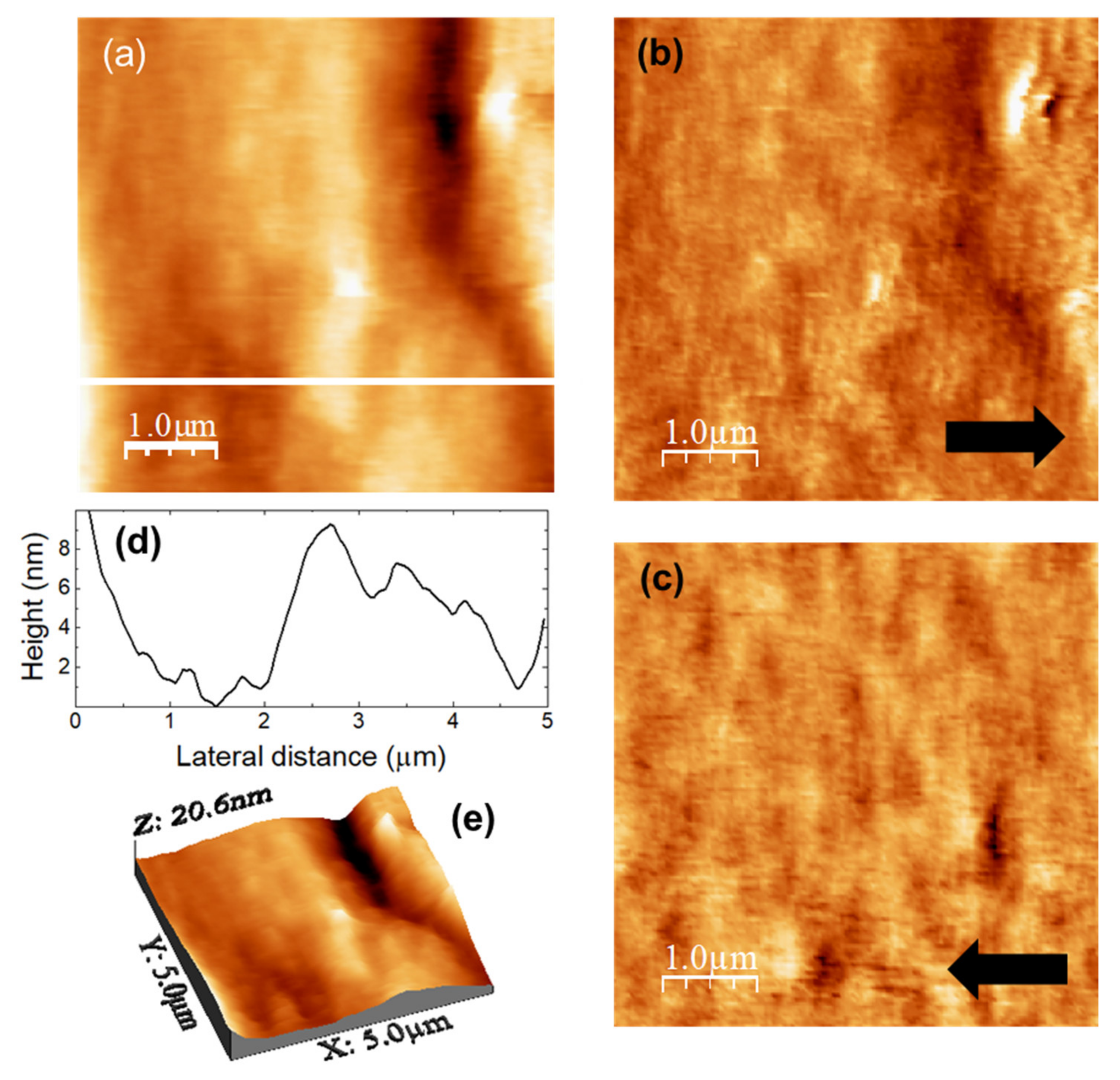
The images in
Figure 4 were recorded over a (5000 × 5000) nm surface area of a DMA-loaded u-PEO film.
Figure 4a is the surface topography, and
Figure 4b,c lateral force microscopy (LFM) images scanning from right to left (
Figure 4b, recorded simultaneously with
Figure 4a) and from left to right (
Figure 4c, each line recorded as the tip travelled back along the line when recording
Figure 4b).
Figure 4d shows a height-contour profile along the continuous white line in
Figure 4a, and
Figure 4e is a 3D representation of
Figure 4a. The surface morphology appears slightly more compact than this of the unloaded u-PEO500 film (
Figure 3a,e). In
Figure 4a,e we may observe a groove, and
Figure 4d shows that the areas without groove exhibit a corrugation a bit larger than those of the areas without pores on the unloaded u-PEO surface (
Figure 3c). No traces of the presence of DMA can be distinguished on the film surface. The absence of frictional contrast in the LFM images
Figure 4b,c confirms the chemical homogeneity of the film surface.
In contrast, on DMA-loaded u-PPO films’ surfaces the coexistence of different species or domains is apparent. The images in
Figure 5a,b were simultaneously recorded over a (5000 × 5000) nm surface area.
Figure 5a is the surface topography, and
Figure 5b is the ultrasonic force microscopy (UFM) image, which allows us to distinguish nanoscale regions with different stiffness and/or adhesion [
7,
32]. The surface morphology in
Figure 5a is markedly different from that of the unloaded u-PPO surface (
Figure 3b); after loading with DMA, clusters of different sizes can be distinguished on the film surface. The largest clusters in this image, marked with dashed circles in
Figure 5a,b are ~300 nm in diameter, and ~80 nm in height. In the UFM image (
Figure 5b), such clusters give rise to a brighter contrast, indicative of a higher stiffness. Interestingly, in UFM, sample regions with distinctly darker contrast are clearly noticeable. Even though, in principle, darker contrast in UFM should indicate softer areas, in polymer nanocomposites, depending on the filler/matrix interface properties, the locations of the filler appear with a darker contrast in UFM due to ultrasound scattering at the interface regions [
32]. The morphology in those regions (
Figure 5a) is characterized by rounded clusters or dots of ~100 nm in diameter, which appear aligned along a well-defined direction, indicated by a dashed white arrow in
Figure 5a.
Figure 5c,d are LFM images recorded over the same surface area than
Figure 5a,b, immediately after these, after suppression of the ultrasonic excitation. In this case, it is clearly noticeable that in the regions in which the UFM contrast appeared darker, such as those between the two dashed white lines outlined in
Figure 5c,d, the surface dots in the LFM images exhibit different frictional contrast, with lower friction then their surroundings (LFM image contrast darker when scanning from right to left (
Figure 5c), and brighter when scanning from left to right (
Figure 5d)).
Figure 5e shows a contact-mode topographic image recorded on a different area which also shows the tendency of the dots to align along a well-defined direction; in this image, the coalescence of some dots into segments along that direction is even clearer than in the area in
Figure 5a.
Figure 5f displays a height contour profile along the continuous white line in
Figure 5e, showing that the dots are here smaller than ~200 nm in diameter, and 40 nm in height.
Figure 5g correspond to a (25 × 25) µm topographic image recorded over a different surface area of the DMA-loaded u-PPO film. Here, the surface morphology unequivocally reveals the formation of a needle-shaped crystal within the u-PPO film, as a result of the incorporation of DMA. DMA crystalizes in the orthorhombic crystal, and the needle-like crystal in
Figure 5g exhibit planes with angles of 90°, characteristics of the orthorhombic morphology. Hence, according to the information provided by the applied AFM techniques, the incorporation of DMA molecules in the u-PPO precursor solution results, after the completion of the sol-gel process, mostly, in the formation of small, rounded clusters or dots, differentiated from the matrix film, which tend to be aligned along a well-defined direction, and, also, in the occasional formation of bigger apparently crystalline needle-like clusters oriented along the same direction.
DMA has a great tendency to form solvents [
33]. Two anhydrous polymorphic forms (Form I and II) are known, and two different monohydrated polymorphs have been reported (DEX I and DEX II) [
34,
35]. In addition, DMA presents an original behavior during its crystallization in specific conditions, and the formation of whisker-like DMA crystals that very much resemble the needle-like crystal observed in
Figure 5g corresponding to a sesquihydrated form, has also been reported in the literature, for example, when saturated ethanolic solution of DMA is injected in water, or when DMA crystallized as dimethylsulfoxide (DMSO) solvate is immersed in water [
36,
37,
38]. The mechanism of formation of these whiskers is not yet fully understood, but it is believed to involve a high local saturation that leads to precipitation with dendritic growth. During the sol-gel process, the solvent evaporates while water is formed during the condensation/hydrolysis reactions of the precursors’ molecules, so it is plausible that the formation of the needle-like crystals in
Figure 5g originates similarly to the aforementioned DMA whiskers. The observation of these crystals in u-PPO400 is of high interest [
39].
DMA has previously been incorporated into bioresorbable films of poly(lactic acid) (PLA) and copolymers of lactic acid and glycolic acid (poly(DL-lactic-co-glycolic acid), (PDLGA)), and the morphology of the loaded films has been studied in detail [
16,
17]. The films were prepared by solution casting, with the polymer dissolved in a solvent and mixed with the drug prior to casting, followed by isothermal heat treatment after preparation. The DMA location/dispersion in the film was controlled by considering solubility effects in the starting solution and the solvent evaporation rate, which determined the kinetics of drug and polymer solidification. For high drug concentrations in the initial solution, and fast solvent evaporation rates, films with small drug particles and crystals were obtained, with drug nucleation and segregation, as well as merging of the small DMA particles to form larger crystals occurring within the dense polymer solution. In ureasil-polyether films prepared by the sol-gel procedure, the film time formation, and therefore the solvent evaporation time, is relatively fast, faster for unloaded u-PPO than for u-PEO, of the order of ~200 s for precursor molecular weights of 2000 and 1900 g·mol
−1, respectively [
40], and the DMA molecules could behave similarly, allowing us to explain the origin of the small dots observed in DMA-loaded u-PPO films, and their apparent coalescence into segments, noticeable, for instance, in
Figure 5e.
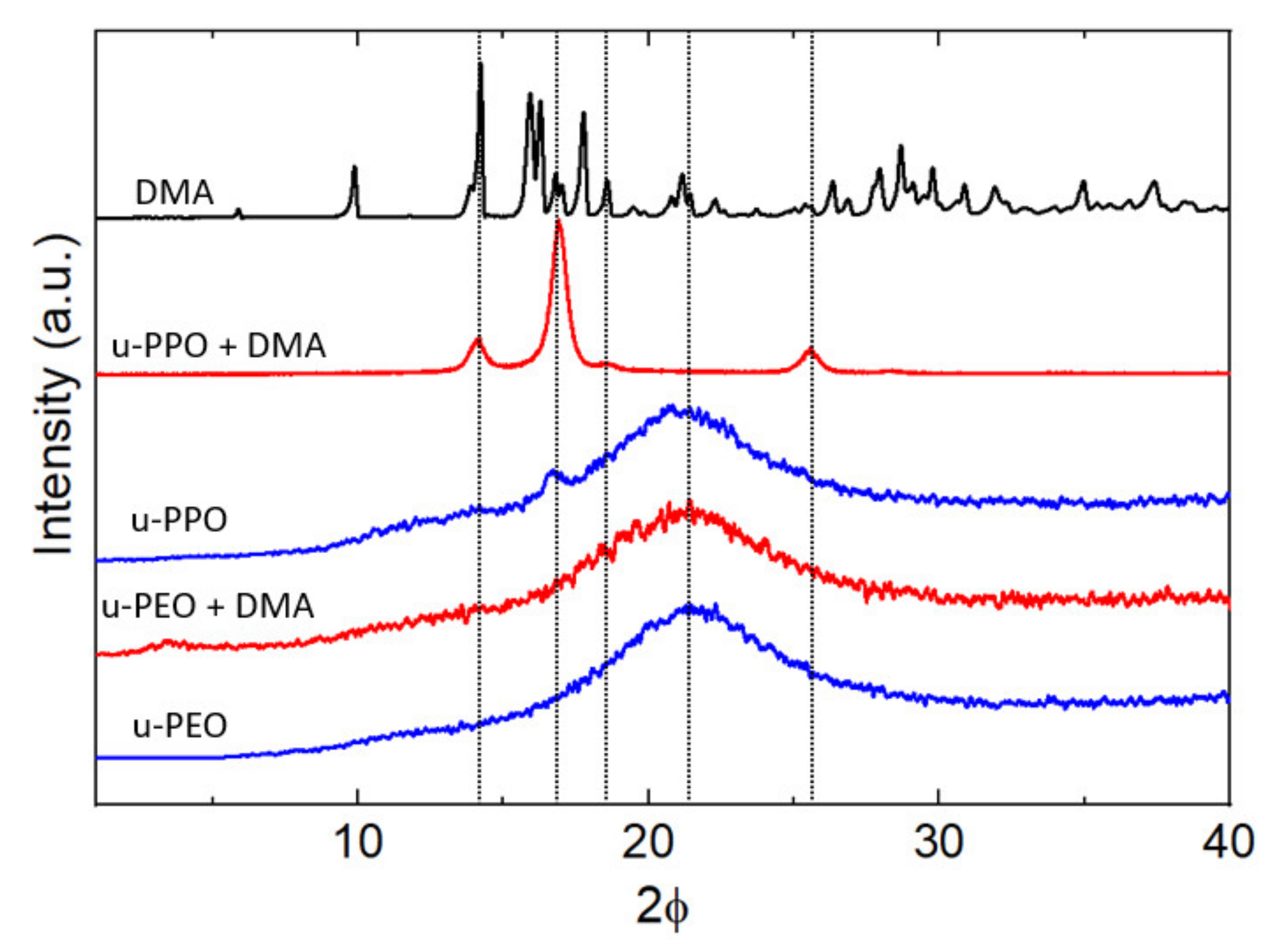
Figure 6 shows the XRD data recorded on unloaded (blue curves) and DMA-loaded (red curves) u-PEO500 and u-PPO400 films, as well as on the original DMA powders (top black curve). Both unloaded u-PEO and u-PPO films exhibit a broad peak characteristic of amorphous materials with its maximum located at 2θ = 21.6° in u-POE, and 2θ = 21.4° in u-PPO. Interestingly, an additional small peak is apparent in the unloaded u-PPO spectra, located at 2θ = 16.8°, which indicates a small amount of crystallinity. This peak corresponds to an interplanar spacing of 5.2 Å, which coincides with the 200 reflection of the orthorhombic structure of crystalline PPO [
41,
42]. However, for u-PPO400, the PPO moieties in the precursor molecules are quite short (see
Figure 1, n < 7 for the u-PPO precursor), and not necessarily isotactic. This result is quite surprising, since to our best knowledge, even though in some cases coexistence of crystalline and amorphous phases has been observed, for instance, in u-PEO samples obtained from high molecular weight precursors (of molecular weight around 1000 g/mol or higher) [
10], no trace of a crystalline phase has been previously observed in ureasil-polyether films obtained from low molecular weight precursors.
The diffractogram of the DMA-loaded u-PEO film does not experience any significant modification with respect to that of the unloaded u-PEO. In contrast, in the DMA-loaded u-PPO sample, the data clearly reveal the presence of a crystalline phase.
Table 1 lists the reflections observed in the DMA-loaded u-PEO diffractogram, the corresponding interplanar spacing, and the crystallite size, derived from Debye-Scherrer equation.
It is noticeable from
Figure 6 that the peak at 16.8° on the unloaded u-PPO matrix is increased in the DMA-loaded film, and slightly shifted to 16.9°. It happens that this peak is coincident with a reflection from the monohydrated DMA form (DEX I) [
34,
36]. The peak at 14.0° corresponds to most intense peak in the original DMA powder diffractogram (top black curve in
Figure 6) and can be found as a strong peak in the diffractogram of the DMA anhydrous variety (FORM II) [
36], but not in this one of the monohydrated phase. Regarding the peak at 25.5°, it is coincident with the 210 reflection of the orthorhombic structure of crystalline PPO [
41,
42], and it is not present as a strong peak in either the anhydrous or the monohydrated DMA phases. Hence, in view of the experimental data, we cannot reject the possibility that a crystalline PPO phase forms within the u-PPO matrix, enhanced by the presence of DMA, which also appears to be present in crystalline state.
At first sight, it is surprising that the peaks of the sesquihydrate phase of DMA, which should correspond to the needle-like crystals in
Figure 5g, considering its resemblance to the DMA whiskers, do not appear in the XRD diagram. We attribute this to the fact that this crystal phase is forming in small quantity, and just at the sample surface, while the information recorded in XRD comes from a surface region of several microns.
For the application of the Debye-Scherrer equation that allows us to estimate the crystallite sizes of the present phases we have considered the dimensionless shape factor K as 0.94, i.e., presuming that the domains have rounded shape. According to the morphology in
Figure 5a,e this should indeed be the case. The obtained data of ~27 nm for the crystallite size is also consistent with the AFM images in
Figure 5. Even though the largest clusters in
Figure 5a reach up to ~300 nm, and in
Figure 5e ~100 nm, there are many smaller dots of ~30 nm in size. Furthermore, the ones that we may appreciate in the image are located on the film surface, and it is to be expected that the ones located below are probably somewhat smaller, being constrained by the surrounding matrix.
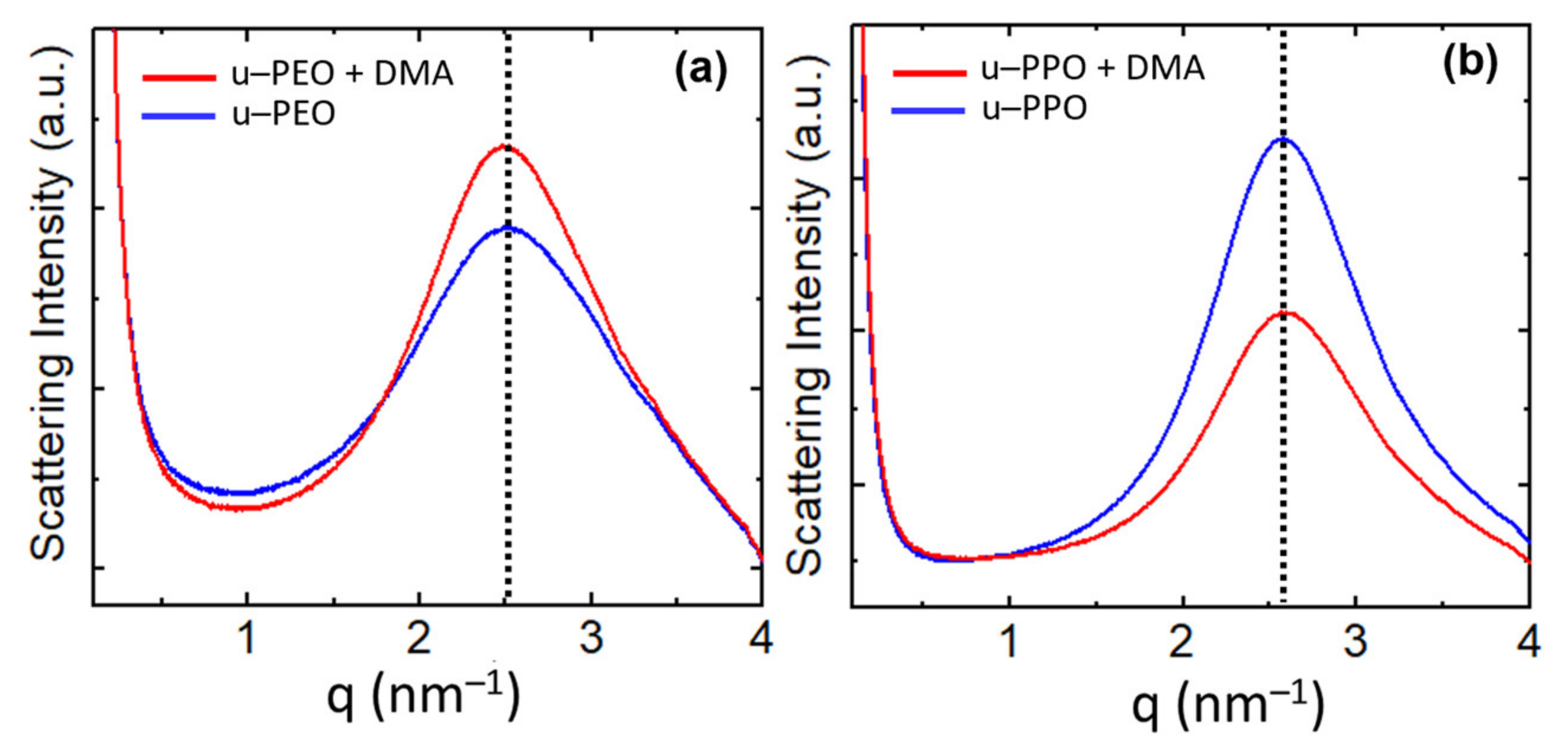
Figure 7 shows the SAXS curves recorded on (a) u-PEO500 and (b) u-POP400. The blue curves correspond to unloaded samples and the red to samples loaded with DMA. The SAXS intensity, I(q), is proportional to the Fourier transform of the correlation function of the electronic density of the material. The peaks give evidence of a strong spatial correlation between the ureasil-polyether cross-linked siloxane nodes. For both unloaded u-PEO and u-PPO the maximum peak position is located at
, and the averaged most probable distances between two adjacent siloxane nodes within the polymer matrix can be estimated by
, where
qmax is the modulus of the scattering vector at the peak’s maximum. As seen in
Figure 7, the introduction of DEXA does not significantly alter this distance, the maxima of the red curves in
Figure 7a,b remain approximately in the same position as those of the blue ones. An average size of the correlation volume associated with the spatial distribution of siloxane nodes,
Lc, can be obtained by applying the Scherrer equation in the case of low-angle X-ray scattering (
) where Δ
q is the full width at half-maximum of the correlation peak of the SAXS function [
43,
44]. The values of
Lc for the SAXS peaks in
Figure 7 are ~9.0 nm for the unloaded and DMA-loaded u-PEO films, and ~12.5 nm for the unloaded and DMA-loaded u-PPO films.
Interestingly, as DMA is introduced in u-PEO, the intensity of the correlation peak increases, indicating the increase of the electronic-density contrast between the ureasil nodes and the polymeric matrix, as observed in other cases [
1,
43]. On the other hand, in u-PPO films, the intensity decreases.
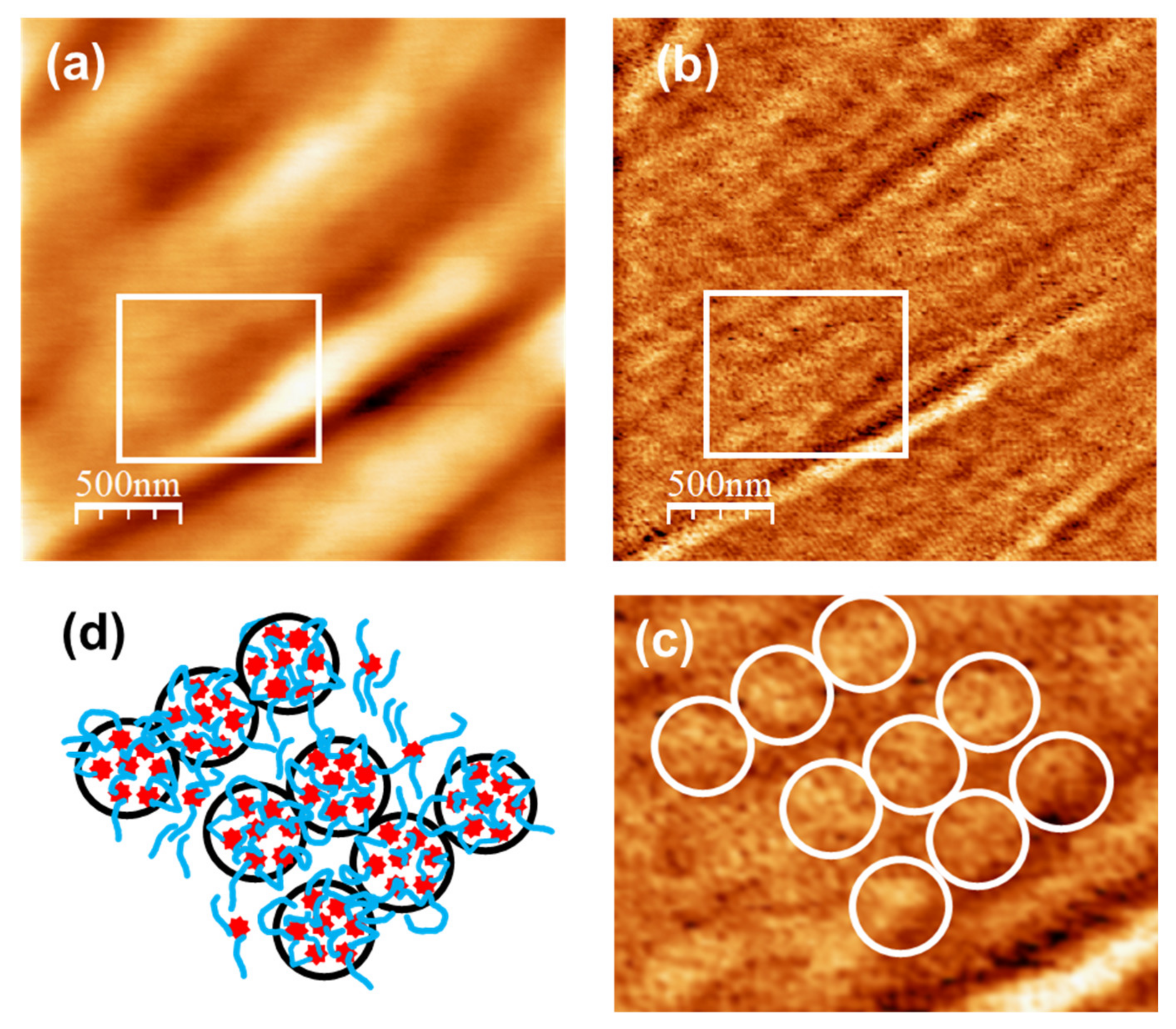
Figure 8a,b corresponds to AFM and UFM images recorded over a (2500 × 2500) nm surface area on the unloaded u-PPO film.
Figure 8a is the surface topography recorded in contact-mode and
Figure 8b is the UFM image recorded simultaneously with
Figure 8a, over the same surface area. As
Figure 3b,f,
Figure 8a shows that the u-PPO400 matrix is formed by polymer ropes, oriented along a well-defined direction. The UFM image (
Figure 8b) makes it possible to distinguish that the ropes are formed by “strands” ~180 nm in diameter, which in turn are structured in rounded clusters.
Figure 8c is a crop of the area delimited by the white rectangle in
Figure 8b, in which some rounded clusters have been enclosed by white circles to facilitate their identification. The clusters yield a brighter contrast in UFM, which indicates that they correspond to stiffer regions. The contrast in
Figure 8b can be understood if the siloxane nodes within the u-PPO400 matrix assemble into “hybrid clusters”, ~180 nm in diameter, corresponding to the rounded clusters seen in the image. In fact, according to the information provided by SAXS (
Figure 7), each of these clusters should be formed by ~14 (i.e. (cluster diameter)/
Lc) disordered hybrid “supercrystals” defined by aggregates of siloxane nodes with average separation distances of ~2.5 nm between them.
Figure 8d depicts a tentative sketch of the u-PPO400 matrix structure, in which the red stars represent the hybrid supercrystals of siloxane nodes aggregates that give rise to the SAXS correlation peak in
Figure 7b.
The model in
Figure 8d provides insight into the results of
Figure 5. The new crystalline phases formed within the DMA-loaded u-PPO400 matrix may occupy the sites in-between the strands defined by the aligned clusters in
Figure 8d. Furthermore, the “stiffer clusters” observed on the surface of the DMA-loaded u-PPO400, (those enclosed with dashed circles in
Figure 5a,b), surely correspond to some of the clusters in
Figure 8b, displaced from their sites as the new phases formed upon drug loading.
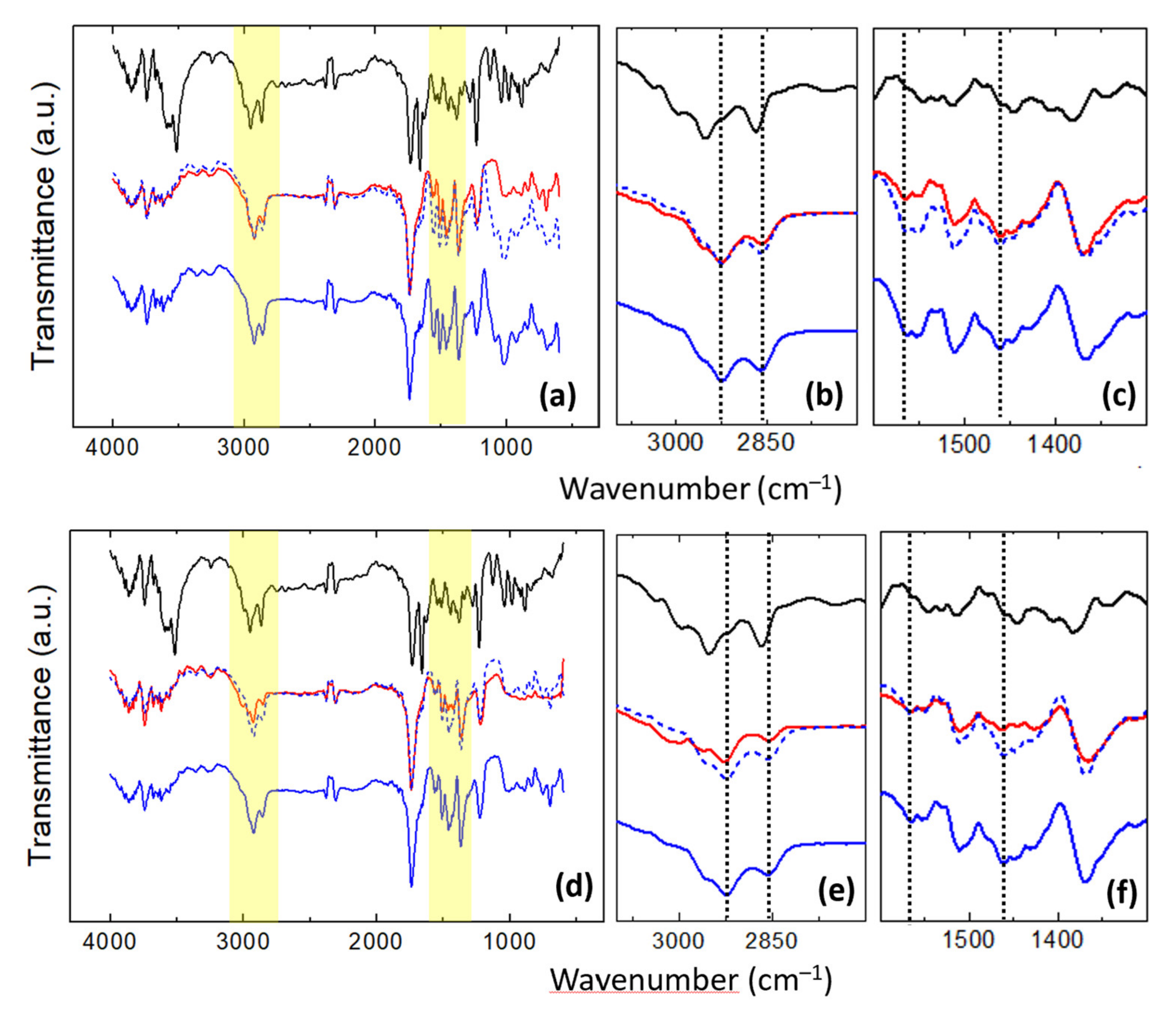
Figure 9 displays the FT-IR spectra recorded on unloaded (blue curves) and DMA-loaded (red curves) (
Figure 9a–c) u-PEO and (
Figure 9d–f) u-PPO films. The black curves in
Figure 9 correspond to the FT-IR spectrum recorded on the original DMA powders.
Table 2 lists the main vibrational peaks observed in the FT-IR spectra and their assignments [
4,
34,
45]. Due to the similarity of the functional groups, most of the characteristic vibrational bands of the ureasil-polyether matrix and the DMA molecules occur in the same regions.
For the discussion of the information provided by the FT-IR data we will mainly focus on the yellow regions in
Figure 9a,d enlarged in
Figure 9b,c,e,f.
Figure 9b,e shows the C-H stretching region. Here, in both unloaded u-PEO and u-PPO curves (blue curves in
Figure 9b,e), the peaks at 2925 cm
−1 and 2859 cm
−1, assigned to CH
2 antisymmetric and symmetric stretching, respectively [
4], have been indicated with a vertical dashed line. In the original DMA powder spectrum (black curves in
Figure 9b,e), we encounter several bands in this region corresponding to olefinic and aliphatic CH stretching [
45]. In DMA-loaded u-PEO films, the incorporation of DMA to the matrix does not bring much change in the FT-IR bands (see red curve in
Figure 9b). Nevertheless, in DMA-loaded u-PPO films, the incorporation of DMA is accompanied by a reduction in the CH
2 stretching vibrations observed in the unloaded samples, and by the emergence of a small peak at 3020 cm
−1 which can be related to olefinic CH stretching from the DMA molecules (see red curve in
Figure 9e). The fact that the CH
2 stretching vibrations diminish confirm that the u-PPO structure is being altered in the presence of DMA, which is in agreement with the results obtained from the AFM and XRD studies.
Figure 9c,f includes the amide and CH
2/CH
3 absorption bands. The peak at 1564 cm
−1 indicated by a vertical dashed line, discernible in both spectra of the unloaded u-PEO and u-PPO films (see blue curves in
Figure 9c,f), has been previously assigned to the amide II band [
4]. The amide II mode is a mixed contribution of the N-H in-plane bending, the C-N stretching and the C-C stretching vibrations, and it is very sensitive to both chain conformation and intermolecular hydrogen bonding. As it is well known, the urea compounds have a very strong self-association capacity, and in small molecular weight ureasil-polyether, the formation of many urea-urea linkages is expected. The N-H groups of the urea moieties are prone to form strong hydrogen bonds with either the carbonyls of the urea moieties of another molecules, and/or with the ether oxygens of the polyether moieties, capable to act as hydrogen bond acceptors. Interestingly, in DMA-loaded u-PEO film this peak strongly diminishes (
Figure 9c), indicating that the presence of DMA in u-PEO alters the interactions of the N-H that exist in the unloaded matrix.
Consistent with our previous observation, the peak at 1100 cm
−1 in the u-PEO film (
Figure 9a, blue curve) which falls into the COC stretching region [
46], strongly diminishes when DMA is incorporated (
Figure 9a, red curve). In contrast, the amide II band does not experience any changes in u-PPO films when they are loaded with DMA (
Figure 9f). In u-PPO, due to the presence of the additional CH
3 group, it is conceivable that the DMA molecules cannot easily access the ether oxygens of the polypropylene moieties. DMA may interact with the u-PPO matrix at sites near the ureasil nodes but if the DMA molecules reach the sites amidst the strands of clusters in
Figure 8, where there are no ureasil nodes, they are bound to interact with each other and form the new phases observed by both XRD and AFM. The fact that u-PPO is structured in strands formed by aligned clusters which consist in aggregates of siloxane-nodes, as illustrated in
Figure 8, explains that the new DMA phases form along a well-oriented direction, growing between the cluster strands.
The peak at 1460 cm
−1, also marked with a vertical dashed line and noticeable in both spectra of the unloaded u-PEO and u-PPO films (blue curves in
Figure 9c,f) is assigned to a C-H mode. Consistent with the results discussed in relation to
Figure 9b,e, this band experience no changes when u-PEO is loaded with DMA, but it is severely affected (the intensity of the band is reduced, i.e., the vibrational modes are hindered) when DMA is incorporated into the u-PPO films.
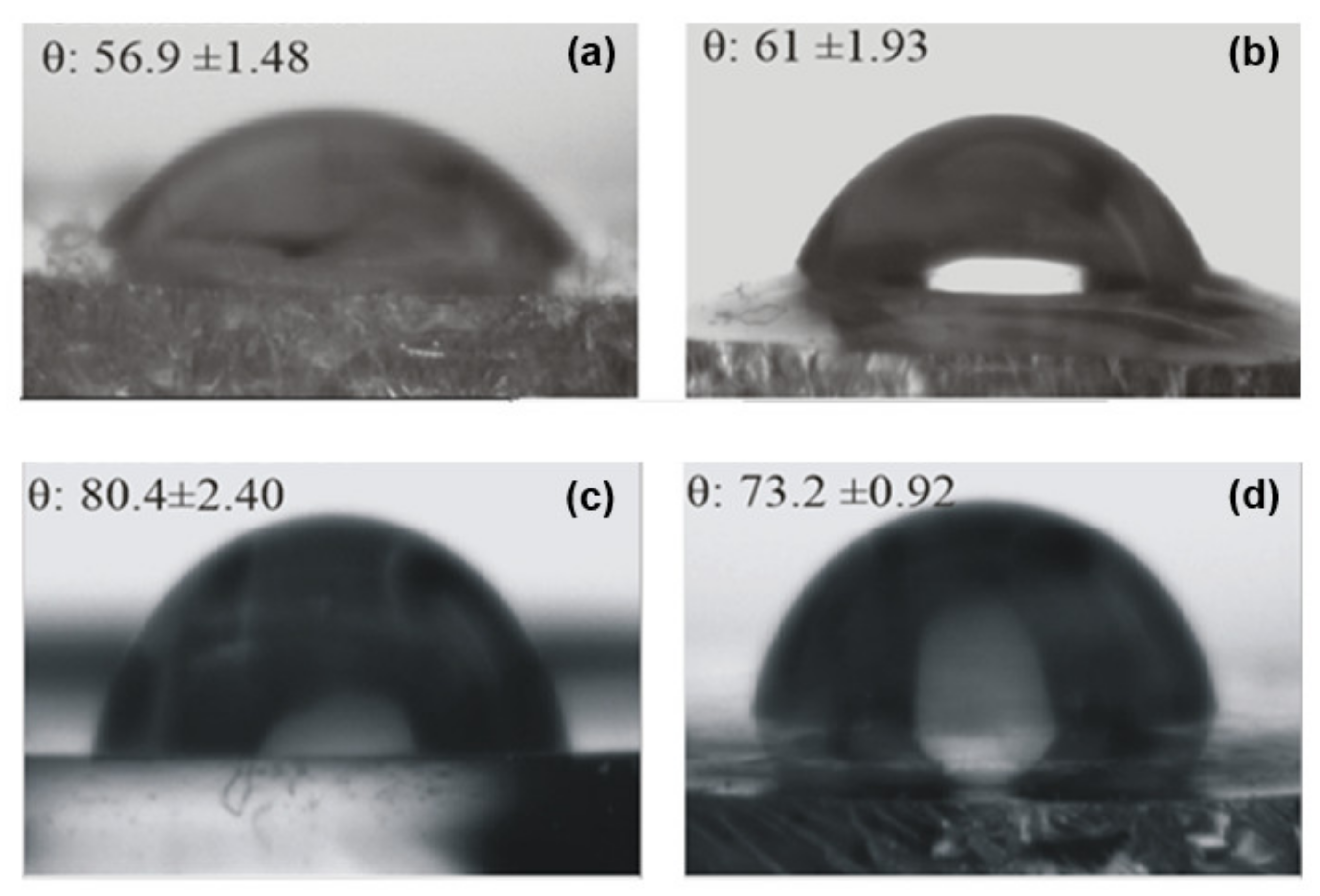
Figure 10 presents the values obtained from contact-angle measurements (θ) on the ureasil-polyether films’ surfaces, together with the recorded optical images. The comparison of the contact angle of the unloaded u-PEO500 and u-PPO400 films indicates that the value is significantly larger for u-PPO, which is consistent with its higher hydrophobic character. Due to its higher hydrophobic nature, u-PPO has lower affinity with water and therefore exhibits a higher contact-angle value. The incorporation of DMA induces changes in the contact angle values in both ureasil-polyether films.
In the DMA-loaded u-PEO500 films, the contact angle increases as a result of the addition of DMA. According to the AFM results (
Figure 4), no traces of DMA are found on the loaded u-PEO films surfaces. Nevertheless, as indicated by FT-IR (
Figure 9), DMA does cause modifications in the u-PEO matrix by hindering the interactions between amide and ether oxygen from different polymer chains. If DMA and the poly(oxyethylene) moieties form complexes involving the ether oxygen, the presence of DMA may facilitate the elimination of water produced during the sol-gel reactions outside the hybrid film and reduce the number of reactive sites at surface locations in such a way that the resulting film surface exhibits reduced hydrophilicity.
In the case of u-PPO400 films, the addition of DMA to the polymer matrix diminishes the contact-angle value (i.e., increases the surface hydrophilicity). DMA is considered a hydrophobic molecule, with a low solubility in water. Nevertheless, the result is understandable considering that the structure of the u-PPO film is severely disrupted in the presence of DMA, with the formation of new crystalline phases. The increased hydrophilicity in DMA-loaded u-PPO400 film is bound to result in an increased bioadhesion of the film, with an increased probability of H-bonds formation on the film surface [
13].
To evaluate surface modifications as a result of DMA release from u-PPO films, we immersed the samples in a solution containing 500 mL of medium (phosphate buffer 7.2 pH with 0.5% of procetyl AWS
® (CRODA, Rawcliffe Bridge, UK)) in a manner similar to the drug release experiments reported in
Figure 2, for 24 h, to ensure that little or no drug remained to be released. The images in
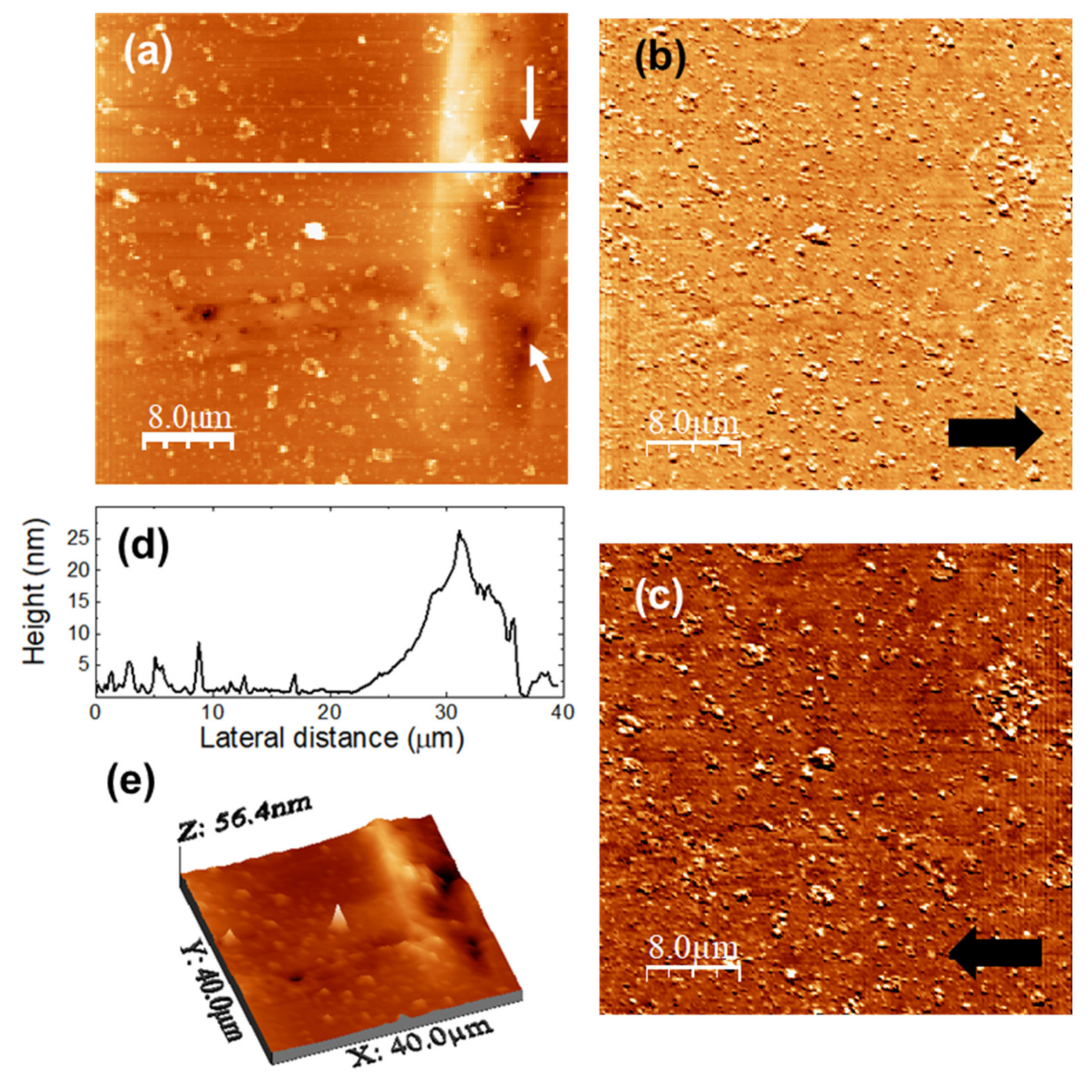
Figure 11 were recorded over a (40,000 × 40,000) nm surface area of the u-PPO film after release of DMA into the medium.
Figure 11a is the surface topography, and
Figure 11b,c lateral force microscopy (LFM) images scanning from right to left (
Figure 11b, recorded simultaneously with
Figure 11a) and from left to right (
Figure 11c, each line recorded as the tip travelled back along the line when recording
Figure 11b).
Figure 11d shows a height-contour profile along the continuous white line in
Figure 11a.
Figure 11e is a 3D representation of
Figure 11a. The absence of frictional contrast in the LFM images
Figure 11b,c confirms the chemical homogeneity of the film surface, i.e., the absence of DMA-related clusters on the film surface.
The surface morphology supports the information provided by the analysis of the release kinetics (
Figure 2), according to which DMA release from u-PPO matrices mostly occurs by Fickian diffusion. No sign of erosion or swelling of the matrix substrate is apparent. The morphology is characterized by the presence of small clusters, typically lower than ~10 nm in high. The ~20 nm rectangular protrusion on the right-hand side of the image clearly indicates the previous location of a DMA crystal. Precisely at the protrusion sides, some small pores are apparent, such as those indicated by the white arrows in
Figure 11a, also clearly noticeable in
Figure 11e, which presumably form as the drug is released. Apparently, the molecular chains of the polymer matrix accompany the drug and re-form, as the drug diffuses outwards, leaving the matrix substrate.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}